SCIENTIFIC COMMUNIQUÉ VIII
- ASX: CUV
- Frankfurt Börse: UR9
- Level 1: CLVLY
DNA Repair Mechanisms
December 2020
Introduction
DNA is the repository of genetic information, often referred to as the biological blueprint for all living cells. Collectively the human genome is composed of 30,000 genes, with each gene carrying the information needed to produce, or synthesise, different proteins. With proteins required for almost all tasks of cellular life, preserving the integrity of our genes, and therefore DNA, is thus critical to upholding normal cell activity and survival. More importantly, all existence relies on intact and unchanged DNA being replicated and transmitted from a cell to its next generation. This allows for the normal growth and development of the organism itself, and the production of healthy offspring.
Ultraviolet radiation (UVR) and high energy visible (HEV) light are potent mutagens (see SCIENTIFIC COMMUNIQUÉ VI). These energetic agents react with various molecules found within our skin cells including our DNA. The damage caused to the structure of our DNA is known as a lesion, with hundreds of thousands generated per hour of sun/UVR exposure. Without an appropriate response from the affected cells, these lesions are permanently retained within our genome and are mutagenic, threatening overall health and survival. Put simply, DNA mutations the primary cause of all skin cancers. Fortunately, in response to UVR and HEV light assault, DNA damage responses (DDRs) are upregulated in our skin cells. DDRs are a combination of diverse cellular signalling cascades which identify the presence of a DNA lesion and subsequently help coordinate its removal. The most favourable DDR upregulates DNA repair. In this COMMUNIQUÉ we will first examine the mechanisms behind different DNA repair systems, then discuss the consequences of defective DNA repair in the skin.
*This communique will build on the knowledge detailed in SCIENTIFIC COMMUNIQUÉ VI.
DNA Repair Mechanisms – Overview
The presence of cellular DNA repair mechanisms was first accidentally observed in the 1940s. Following the discovery of penicillin, the biologist Albert Kelner spent his post-doctorate attempting to artificially manipulate the bacteria Streptomyces griseus, eager to generate mutant strains that would produce alternative and (ideally) more effective antibiotics. At the time, the gold standard in mutation induction was through UV irradiation. However, when subjected to high doses, Kelner’s cultures rapidly disintegrated, frustrating his experiments. Through stubborn driven persistence, he discovered that the survival of these microorganisms could be reinstated by placing his cultures under fluorescent lamps. Simultaneously, Italian virologist Renato Dulbecco was examining the progress of bacteriophage replication in the presence of UV light. By chance, he noticed the discarded cultures that sat by his desk light outlived those thrown in light deprived areas. Both scientists had independently identified the same repair system in microorganisms: enzymatic photoreactivation (EPR). EPR was found to be a light dependent recovering mechanism undertaken by these prokaryotic cells to restore the damage to their DNA following UVR exposure. The molecules which facilitated this repair were photoactivated by wavelengths of light only seen in the visible spectrum, explaining why instead of lethality, survival and retained viability was seen in these treated cell cultures.
The effects of radiation on organisms was an area of biology that had already started to receive much attention after the emergence of atomic weapons. This, coupled with the proof of DNA specific repair systems in primitive organisms by Kelner and Dulbecca, accelerated the discovery of further repair mechanisms that excise DNA damage caused by genotoxic agents to restore genome integrity.
It is now known that evolutionary conserved DNA repair mechanisms are active in all organisms, including humans. Roughly 2% of the human genome encodes for DNA repair molecules which take part in one of five known mammalian DNA repair pathways (Table 1). Each of these processes require a unique combination of DNA repair proteins that interact with one another in a sequential manner to identify, remove and repair any acquired damage within our DNA.
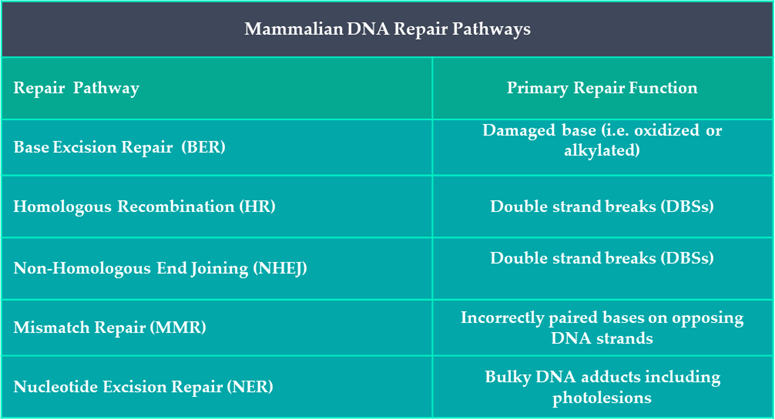
Table 1: Summary of DNA Repair Pathways in Humans.
A DNA lesion can be defined as anything from a base modification to a strand break. After solar radiation exposure, our skin’s DNA has acquired three predictable types of lesions: oxidised bases; strand breaks; and the intralinking of pyrimidine containing nucleotides (see SCIENTIFIC COMMUNIQUE VI). Different repair mechanisms are upregulated in the presence of different lesions. Examining the cells that compose our skin’s epidermal and dermal layers has illustrated the sudden increase of four distinctive DNA repair mechanisms after sun exposure: nucleotide excision repair (NER); base excision repair (BER); Homologous Recombination (HR); and Non-Homologous DNA end Jjoining (NHEJ). Activation of DNA repair mechanisms are the most favourable cellular outcome following damage as they promote the reparation and survival of functional cells. Yet the absence of any one of these four DNA repair pathways can have serious health consequences.
Nucleotide Excision Repair (NER)
NER is the specialised DNA repair pathway for large helix distorting DNA lesions, including those caused by UVB (290-320nm). This mechanism allows the efficient removal of photoinduced cyclobutane pyrimidine dimers (CPDs) and pyrimidine 6-4 pyrimidone photoproducts (6-4PPs), subsequently reinstating the stability of cellular DNA prior to mutagenesis (see SCIENTIFIC COMMUNIQUÉ VI). The NER mechanism can be divided into two sub-pathways differentiated by the method of DNA lesion identification, Transcription-coupled repair (TCR) and global genome repair (GGR). As suggested by its name, TCR repairs UVB photolesions that have been identified during the transcription of messenger RNA (mRNA), an intermediate molecule that is a copy of a DNA’s gene, required to instruct the synthesise of specific proteins. Under normal circumstances, after gene activation, the enzyme complex RNA polymerase locally binds and unwinds the cell’s DNA where it subsequently moves step wise along the desired nucleotide sequence, generating a complementary mRNA strand to the activated gene. However, following UVB exposure, the bulky size of CDPs and 6-4PPs can stall RNA polymerase in its path, inhibiting the continuation of transcription. It is this obstruction that results in recruitment of additional lesion recognition proteins that indicate to the skin cell the presence of a malformation, which instead fosters the next step in damage removal. However, since not all genes are transcriptionally active, photolesions that have been generated in the genetic sequence of dormant genes or non-coding regions of the DNA must be identified by an alternative approach. In GGR, the DNA is surveyed for any abnormalities by the UV damaging binding complex UV-DDB and XPC-RAD23B. These proteins locate, bind, and tag the DNA distortion, preparing it for removal.
Following lesion recognition, the methodological steps of DNA repair remains identical in both sub-pathways. The DNA helix surrounding the photolesion is first unwound, mediated by the multicomplex protein transcription factor II H (TFIIH). The damage is verified by a protein known as xeroderma pigmentosum A (XPA), which is recruited from the cytoplasm of the skin cell to the nuclear site of damage after DNA unwinding, where it also assists the assembly of the incision proteins, endonucleases. Xeroderma pigmentosum F and G (XPF, XPG) dual cleave the phosphodiester bonds either side of the lesion so that the small section of DNA containing the UVB induced damage is ‘cut’ from the rest of the DNA molecule. The importance of TFIIF’s activity is seen at this point: by unwinding the DNA and holding it open in the correct position, the endonucleases are then able to precisely cleave the DNA sequence containing the lesion only, as opposed to other unaffected areas of the DNA molecule that would otherwise enhance further damage. The segment of excised DNA, or oligonucleotide, is rapidly degraded. In the meantime, DNA polymerase, the enzyme responsible for catalysing the joining of free DNA nucleotides, resynthesises the gap to reform a continuous strand of DNA (Figure 1). This explains why defects in any apparatus of NER can have detrimental phenotypic effects, as seen in xeroderma pigmentosum (XP) patients, as each repair molecule relies on the activity of the previous.
Figure 1: Nucleotide Excision Repair (NER) for bulky photolesions.
TCR (Not shown): The helix distorting photolesion 6-4PP destabilases RNA polymerase II during gene transcription. This triggers the recruitment of Cockayne syndrome protein B and A (CSB and CSA). The subunit of CSA, damage binding complex 1 (DDB1) helps the identification of the lesion. TFIIH is then recruited to the site of the lesion.
GGR: The photolesion 6-4PP is identified by the damage sensor protein complex UV-DDB, which acts as a substrate for the complex’s small subunit XPE (also referred to as DNA damage-binding protein 2, DDB2) which efficiently binds to lesion. The damage recognition complex XPC-HR23B also binds to the non-damage side of the DNA strand where its presence is required for the assembly or other core NER proteins. XPC-HR23B is the essential initial damage recognition factor in GGR. TFIIH is recruited to the site of the lesion and interacts with XPC- HR23B.
TFIIH pries open the DNA molecule around the site of the lesion, with DNA helicases subunits (xeroderma pigmentosum B and xeroderma pigmentosum D, XPB and XPD) unwinding the damaged DNA. When XPB and XPD encounter the lesion, they stall at the site and recruits the precision complex (XPA, RPA, XPG). The damage is verified and XPA recruits and interacts with ERCC1-XPF. RPA aligns ERCC1-XPF and XPG, both of which incises the phosphodiester backbone of the damaged DNA strand 5’ and 3’to the lesion respectively. Initiation of repair synthesis by DNA Polymerase δ and Polymerase ε and associated factors, followed by 3′ incision by XPG. Completion of repair synthesis and sealing of the nick by DNA ligase I completes the process.
Base Excision Repair (BER)

Previous
Next
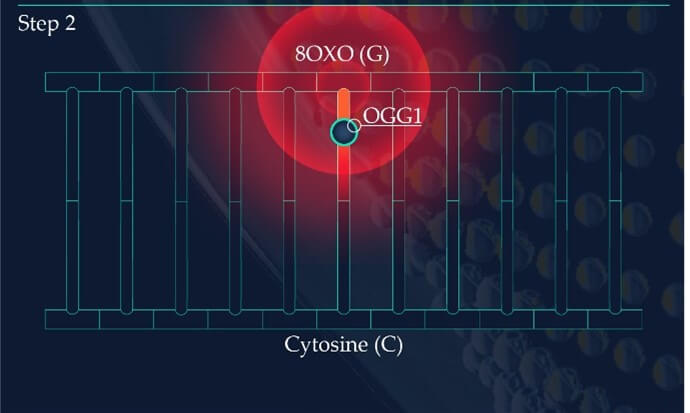
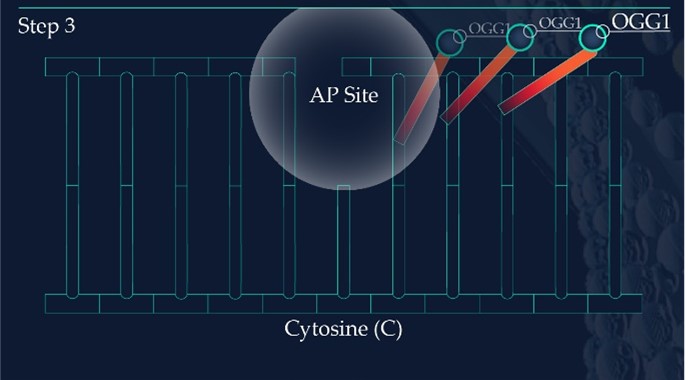
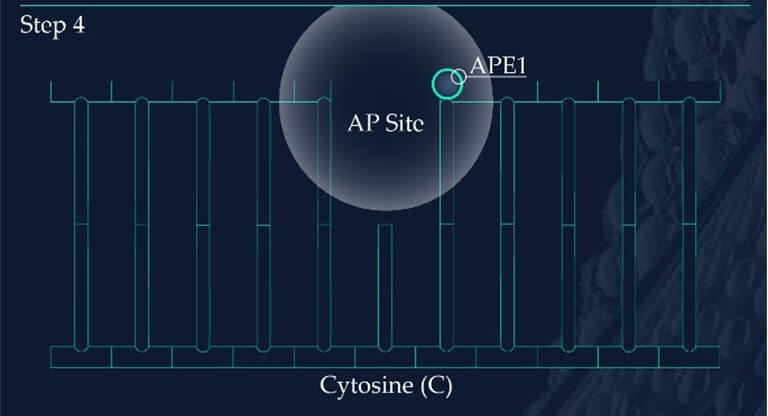
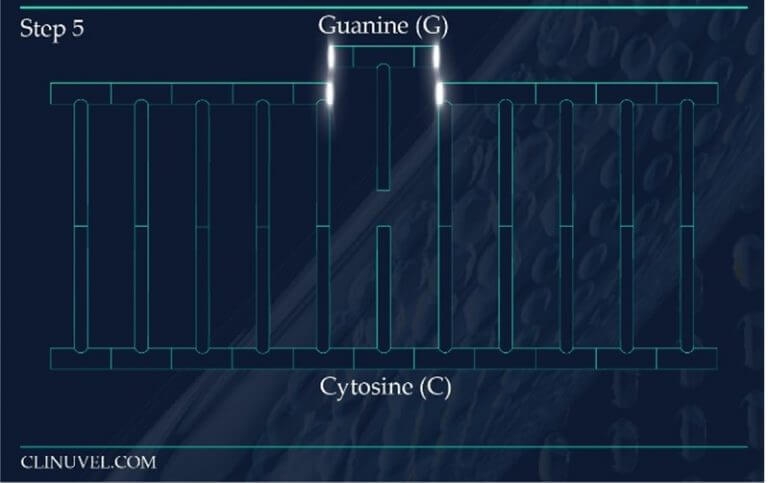
Figure 2: Base Excision Repair.
The DNA glycosylase OGG1 identifies the oxidated base, 8-oxoG, hydrolysing the N-glycosidic bond between the base and the DNA’s phosphodiester backbone. OGG1 also catalyses the hydrolysis of the phosphodiester backbone at the 3’ side adjacent to the damaged base at the same time. This leaves a baseless sugar moiety, the AP site.
Apurinic/apyrimidinic endonuclease 1, APE1, initiates the repair of the AP site by hydrolysing the phosphodiester backbone on the 5’ side immediately adjacent to the damage.
The subsequent synthesis and ligation steps are carried out by polymerase β (POLβ) and X-ray cross-complementing factor 1 (XRCC1), respectively. Polβ is a bi-functional enzyme, catalysing 5′-dRP removal along with gap-filling DNA synthesis. XRCC1 repairs the nick in the phosphodiester backbone to restore an intact DNA molecule.
While NER is the ubiquitous repair pathway for UVB induced adducts, the DNA of skin cells are also subjected to indirect forms of damage that come from exposure to UVR and HEV light. Reactive oxygen species (ROS) serve as a secondary source of DNA damage, generated through the reactivity of photosensitive molecules in the skin with UVR and HEV light. ROS are known to induce the oxidation of DNA nucleotides as explained in SCIENTIFIC COMMUNIQUE VI. The guanine nucleotide base is particularly susceptible to oxidation by ROS, which catalyses guanine to 8-Oxo-7,8-dihydroguanine (8-oxoG). Due to the chemical characteristics of 8-oxoG, this lesion favours base mispairing during DNA replication. 8-oxoG tricks the replication machinery into recognising it as a thymine base as oppose to a guanine base. This is a process which disrupts gene fidelity and is thereby highly mutagenic if left to reside in the skin cell’s genome. DNA repair systems are in place to ensure the removal of abnormal nucleotides which contain this base.
Similar to the detection of CDPs and 6-4PPs, damage sensing molecules are constantly scanning the DNA of skin cells for the presence of oxidised nucleotides. However, if located, the machinery effecting their removal is base excision repair (BER). This differs from NER as it involves the excision of only a single nucleotide base as opposed to larger segments of DNA containing bulky distortions. 8-oxoG presents as a substrate for a specific DNA glycosylase enzyme known as 8‐oxoguanine glycosylase 1 (OGG1). OGG1 is a multifunctional protein which first identifies and then binds to 8-oxoG on the DNA strand, later cleaving the glycosidic bond found between this oxidative base and the backbone of the DNA strand. This results in an abasic site, where a base is now missing from the DNA molecule (Figure 2), referred to as an apurinic/apyrimidinic site (AP site). AP endonucleases such as APE1 cleave and remove the backbone of the DNA at the 5’ end to the AP site. This allows DNA polymerase to resynthesise the replacement nucleotide, with the skin cell’s DNA successfully restored (Figure 2).
DNA Double-Strand Breaks
Highly reactive ROS molecules such as the hydroxyl radicals not only cause the oxidation of DNA bases but can slice the DNA molecule by directly attacking its phosphodiester backbone, leading to a strand break. Strand breaks are discontinuities in the DNA molecule. They are classed as either a single strand break (SSB) or a double strand break (DSB) in which either one or both sides of a DNA molecule’s backbone is severed. DSBs are arguably the most serious and toxic DNA lesion that have disastrous impacts when left unrepaired. They cause the loss of genetic continuity, where daughter cells no longer contain the same number of genes as the parent cell if division occurs. This dramatically effects the progeny cell’s ability to operate as normal, with chromosome abnormalities a hallmark of cancer.
Generally, ROS reactivity initially generates a greater yield of SSBs, without rapid restoration, yet SSBs successively induce DSBs within a DNA molecule. Through a similar method, incompetent NER is also a source for DSBs. In both instances, their generation is consistent with cells housing unrepaired SSBs or photolesions progressing into the S-phase of the cell cycle, alternatively referred to as the DNA replication phase. The standard procedure during duplication involves the DNA replication machinery working to separate the DNA duplex into two component single-strands (ssDNA). In doing so, it creates a ‘chicken foot’ like formation known as the replication fork, with the size of the fork increasing as the process of replication continues (Figure 3). However, when the apparatus encounters one of these remaining lesions, the fork structure collapses with a DBS supervening. Complicating matters further, DSBs also appear after BER and NER endonuclease proteins attempt to excise mutations. Where mutations exist on opposing strands of the DNA less than 10 base pairs from one another, the repair system’s endonucleases create spontaneous DSBs as a result of simultaneous DNA strand expurgation.
Homologous Recombination (HR) is the chief error free restoration mechanism for DSB. This method of DNA repair involves the DSB containing DNA molecule invading a homologous undamaged DNA molecule, such as a sister chromatid (Figure 4). Since both molecules share the same or very similar DNA sequences, the intact sister chromatid can be used as a template for strand repair.

Figure 3: Replication Forks
During DNA replication, the helix of the DNA molecule is opened and DNA polymerase uses the parental DNA strands as templates to transcribe a copy of the cell’s DNA. The replication fork is the area in which the replication of DNA is occurring. A DNA lesion such as a SSB prevents fork progression and instead causes fork collapse. This leads to a DSB.
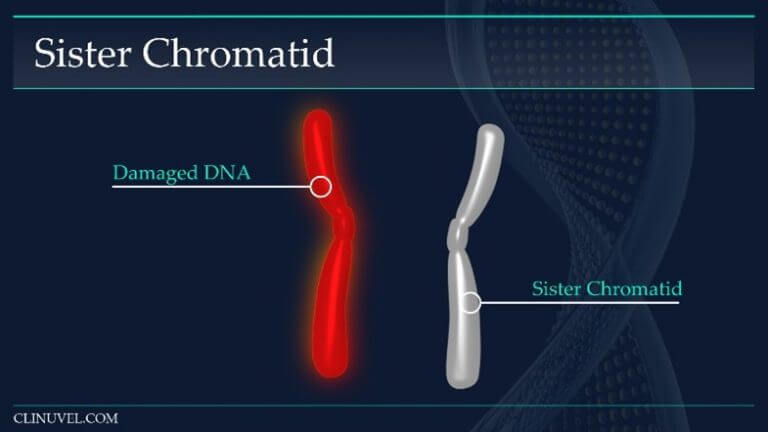
Figure 4: Sister chromatids
They are two identical copies of the same chromosome formed from DNA replication. In HR, a damaged DNA molecule containing a DSB can use the newly synthesised sister chromatid as a template for repair.
Phosphorylated H2A histone family member X (γH2AX) plays a key role in repair initiation. As part of the damage response, γH2AX proteins form as foci around the vicinity of DSBs. γH2AX increases DNA accessibility and the recruitment of DNA repair molecules to facilitate HR. The repair process follows and can be divided into three key events: strand exchange, branch migration and resolution (Figure 5).
While HR is praised as a successful mode of DSB repair, due to the requirement of a homology donor, this repair is only feasible during late S and G2 phases of a cell cycle, after DNA replication has occurred and a resultant identical sister chromatid has been synthesised. Non-Homologous DNA end-joining (NHEJ) is a succeeding and more rapid method of DSB repair with no template requirements, however it is also recognised as a mutagenic form of DNA restoration due to its lack of accuracy. While the additional presence of a secondary DSB repair pathway asserts a survival advantage, in NHEJ the separated ends of DNA are simply “glued” (ligated) together which may result in the loss of nucleotide sequences. This can cause the aberrant alteration of functional genes which may encode proteins that favour the transformation of healthy skin cells into tumorous ones.
https://vimeo.com/487155325/64c4ac2579
Figure 5: Homologous Recombination (HR)
The Cell Cycle and DNA Repair
All the mentioned DNA repair processes take time to occur, however our skin cells are continuously and progressively moving through a cell cycle. The cell cycle enables cells to perform their most basic function: self-replication (Figure 6). This is an ordered sequence of events which mainly involves preparing the cell for division, with its fourth phase (mitosis) resulting in the generation of two identical daughter cells. The rate at which this occurs is highly dependent on the cell type. For example, the time taken to complete one full cell cycle is much shorter in the epidermal basal cells in comparison to the skin’s melanocytes, reflective of the functions of these specialised cells. This is a coordinated process and does not occur passively.
The control of the cell cycle system relies on the presence of proteins which mediate the cell’s transition to the next phase. Cell cycle checkpoint proteins are a class of surveillance serine/threonine-specific protein kinases activated as part of the cell’s DDR. There are numerous cell cycle checkpoint proteins which function to stall the cell at different phases of the cell cycle, with their main goal to ensure that only viable and healthy cells undergo mitosis. This prevents the division of unstable cells harbouring DNA lesions that would otherwise have been propagated to the next generation of skin cells. As part of the cell’s initial DDR, cell cycle checkpoint proteins are upregulated and work in sync with DNA repair mechanisms to fix the structural changes made to the DNA by UVR and HEV light as a means to restore genome stability. Checkpoint kinase 1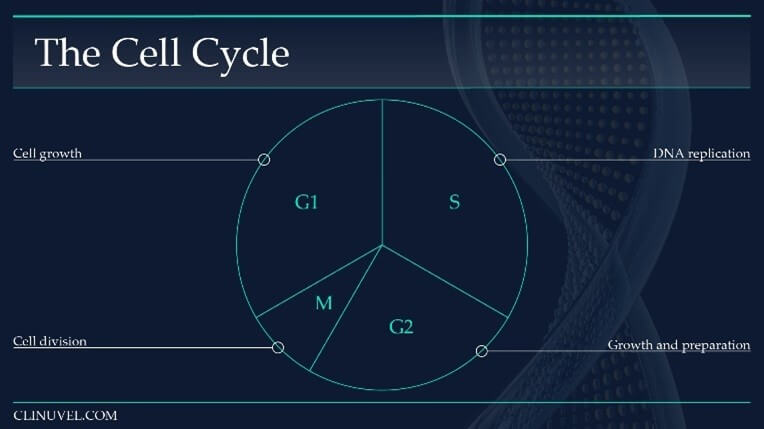
Figure 6: The cell cycle of a somatic cell in eukaryotes (i.e. skin cells).
The cell cycle is a sequence which duplicates a cell’s contents, including its chromosomes, and divides into two. The cell cycle can broadly be divided into four phases, with the length of each phase variable:
Gap1 (G1) Phase: An intermediate phase between the end of cell division (mitosis) and DNA replication (S-phase). The cell grows and its cytoplasmic contents are duplicated. During this phase, a cell decides whether it is capable of accurate DNA replication. If so, it progresses into S-phase.
Synthesise (S) Phase: DNA replication occurs in which the DNA molecule is duplicated.
Gap 2 (G2) Phase: The cell prepares for cell division (mitosis) by synthesising necessary proteins, growing in size.
Mitosis (M) Phase: The cell divides into two cells. The original cell is referred to as the parent cell while the new progeny is a daughter cell. The daughter cell is identical to the parent cell.
(Chk1), for example, temporally halts S/G2 phase transition for skin cells that are in S phase, thus allowing time for repair to occur before the cells prepare for division. Without these checkpoint proteins, DNA repair mechanisms would be competing against biological time restrictions.
Apoptosis and Senescence
It is not always feasible for DNA repair to occur efficiently, especially when the level of damage incurred is recognised as irreparable. Where DNA repair fails, DDR mechanisms must work to restrict the inheritance of this damage to the next generation of skin cells through either premature apoptosis (death) or senescence (irreversible growth arrest). In moderation, these two systems are successful fail safe mechanisms which the cell employs to terminate its ability to divide, either through self-destruction or permanent cell division arrest. However, overstimulation of apoptosis and senescence can also be damaging to our health. This is particularly detrimental to the basal cells, the epidermal stem-like cells of the skin, whose functionality relies on their ability to continuously divide and give rise to keratinocytes, preserving skin thickness. While neither cell fate is more favourable than the repair process, senescent cells remain semi-viable as they retain some metabolic activity, and thereby continue to contribute to skin maintenance. By contrast, excessive apoptosis is associated with tissue atrophy and increased skin vulnerability to both internal and external damage.
For example, dermal fibroblasts synthesise macromolecules that comprise the skin tissue’s extracellular matrix, a mesh network found within the intercellular spaces of tissues. Excessive apoptosis of fibroblasts cells will slow rates of wound healing. In the extreme, this exasperates infections and leads to chronic wound formation associated with loss of functional skin. When frequently occurring in the keratinocytes, the thinning of the epidermis is seen. This compromises the epidermal layer’s ability to function normally. For example, the prevention of excessive transepithelial water loss is no longer as efficient (presenting as dry, rough skin). Abnormal pigmentation will also be characterised in the skin during frequent melanocyte cell death as pigment (eumelanin) distribution is no longer uniform. Eumelanin protects cells against radiation damage, thus poor dispersal leaves areas of the skin vulnerable to radiation damage. Overall, it is recognised that excessive apoptosis contributes to the deterioration of healthy skin, predisposed to further UVR and HEV light damage.
Life Without DNA Repair
Skin tumours develop when the body is unable to cope with the level of damage that is sustained after overexposure to UVR and HEV light. Cells that have acquired mutations as a result are allowed to escape these normal repair or removal pathways as the body simply does not have sufficient resources to inspect each of these cells for damage. Their DNA is not restored efficiently and instead of cell termination, these defective cells continue to survive and divide, increasing the likelihood of skin cancer.
The genetic rare disease XP is a model of defective UVR induced DNA repair. In patients with XP, the DNA repair molecules required to facilitate NER are defective, inhibiting the effectual removal of photolesions. Skin cells, such as the epidermal keratinocytes, containing CPDs and/or 6-4PPs are forced to undergo rapid apoptosis instead, explaining the severe sunburns these patients experience despite minimal sun exposure. However, this also leads to some damaged skin cells avoiding all response pathways. This explains the 10,000 fold increased risk of skin cancer some these patients incur compared to the general population, as skin cells housing oncogenic mutations are instead allowed to thrive and multiply.
While this describes an extreme example of inefficient DNA repair, predisposition to cutaneous malignancies has consistently been linked with variants of the melanocortin 1 receptor (MC1R) gene. This receptor is expressed on the surface of all skin cells including melanocytes, keratinocytes and fibroblasts. The binding of the endogenous ligand alpha-melanocyte stimulating hormone (α-MSH) to MC1R is known to stimulate signalling cascades associated with DNA repair in these cells. Numerous studies now identify increased MC1R activation with enhanced skin cell repair against UVR- and HEV light-induced DNA damage. Evidently in MC1R variant populations, these cascades are less effectual. This explains the geographical trends in skin cancer incidence as over 50% of Europeans have inherited a form of the receptor associated with a reduced function, highlighting a substantial group of individuals at a much higher risk of skin cancer resultant from suboptimal DNA repair (see SCIENTIFIC COMMUNIQUÉ IV).
Summary
DNA repair is crucial to promote the restoration and survival of solar damaged skin cells, and thus our overall skin health. Inefficient DNA repair pathways result in skin cancer, which can be lethal. Apoptosis and senescence are failsafe mechanisms that are upregulated in conditions where DNA repair is not possible. However, in excess, these processes pose their own issues that contribute to declining health. XP is a hereditary consequence of loss of function genetic mutations in the DNA repair molecules that are required to carry out NER, however a large proportion of the global population carry polymorphisms in specific genes that render their skin DNA repair capacity less efficient.
In a future SCIENTIFIC COMMUNIQUÉ, we will focus on intracellular MC1R signalling and its association with DNA repair.
Useful Links
This SCIENTIFIC COMMUNIQUÉ VIII is the second COMMUNIQUÉ in a series of three COMMUNIQUÉs on cutaneous DNA damage and repair in humans. The reader is advised to read all three COMMUNIQUÉs collectively. Links to the preceding COMMUNIQUÉs can be found here:
SCIENTIFIC COMMUNIQUÉ VI – Ultraviolet Radiation Damage and Oxidative Stress in Skin Cancer
SCIENTIFIC COMMUNIQUÉ IX – Beyond Pigment: The Melanocortin 1 Receptor (MC1R) in DNA Repair
Download PDF
Glossary
| Term | Definition |
| 8‐oxoguanine glycosylase 1 (OGG1) | An enzyme which catalyses the first step in the base excision repair pathway (BER) during the repair of 8-oxoG |
| Apurinic/apyrimidinic endonuclease 1 (APE1) | A multifunctional enzyme involved in base excision repair (BER). APE1 cleaves DNA 5′ of an AP site. |
| Apoptosis | Programmed cell death. |
| Apurinic/apyrimidinic site (AP site) | Also known as an abasic site, a location in DNA that has no nucleotide base present. |
| Base excision repair (BER) | A repair mechanism that corrects DNA damage from oxidation, deamination and alkylation. |
| Cell cycle | The cell cycle, or cell-division cycle, is the series of events that take place in a cell that cause it to divide into two daughter cells. |
| Cyclobutane pyrimidine dimers (CPDs) | Molecular lesions formed from thymine or cytosine bases in DNA via photochemical reactions. |
| DNA damage responses (DDRs) | A network of cellular pathways that sense, signal and repair DNA lesions. |
| DNA Lesion | A section of a DNA molecule containing a primary damaged site i.e. a base alteration or a strand break. Replication before repair, or inefficient repair, can result in the fixation of a primary lesion as a permanent mutation. |
| DNA ligase | An enzyme that joins DNA strands together by catalysing the formation of a phosphodiester bond. |
| DNA mutation | Heritable change in the nucleotide sequence of the DNA. |
| DNA polymerase | An enzyme that synthesises DNA by joining nucleotides together using a DNA template as a guide. |
| Distributing arteries | Otherwise referenced as muscular arteries, these are medium sized arteries that transport blood from an elastic artery and branch into resistance vessels. Many layers of smooth muscle cells comprise its tunica media. Examples include the internal carotid arteries. |
| Gene | A unique sequence of nucleotides forming part of a DNA molecule, the order of which is the precursor code for protein(s). |
| Global genome repair (GGR) | A subtype of NER which repairs UVB induced lesions in the untranscribed regions of the DNA. |
| H2A histone family member X (H2AX) | When phosphorylated (γ H2AX), this protein acts as a scaffold for other repair proteins during homologous recombination (HR). |
| Homologous recombination (HR) | A repair process for double strand breaks (DSBs). |
| Melanocortin 1 receptor (MC1R) | A Gs protein coupled receptor and the endogenous target for the hormone alpha melanocyte-stimulating hormone. Activation of this receptors upregulates DNA damage and DNA repair responses. |
| messenger RNA (mRNA) | A single-stranded RNA molecule that is complementary to one of the DNA strands of a gene. It is an RNA version of a gene. |
| MRN complex | A protein complex consisting of Mre11, Rad50 and Nbs1 that takes part in the first stages of homologous recombination (HR). |
| Nonhomologous DNA end joining (NHEJ). | A pathway for the repair of DNA double-strand breaks (DSBs) that can occur anytime throughout the cell cycle. |
| Nucleotide Excision Repair (NER) | A process that repairs damage to one strand of the DNA, particularly from UV irradiation, which distorts the DNA helix. |
| Oligonucleotide | Oligonucleotides are short, single- or double-stranded DNA or RNA molecules. |
| Phosphodiester backbone | This bond is a key structural feature of the backbone of DNA and RNA and links the 3′ carbon of one nucleotide to the 5′ carbon of another. |
| Polymorphisms | Involves one of two or more variants of a particular DNA sequence. |
| Pyrimidine 6-4 pyrimidone photoproducts (6-4PPs) | A DNA mutation formed as a result of UVB radiation exposure. |
| Rad51 | A protein involved in double strand break repair (DSB) in homologous recombination. |
| Reactive oxygen species (ROS) | A reactive chemical species containing oxygen, such as the superoxide anion or hydrogen peroxide. |
| Replication fork | A structure that forms within the long helical DNA during DNA replication. |
| RNA polymerase | Synthesises RNA by following a strand of DNA. RNA polymerase is an enzyme that is responsible for copying a DNA sequence into an RNA sequence, during transcription. |
| Senescence | Irreversible cell cycle arrest driven by a variety of mechanisms, including DNA damage. |
| Strand break | Discontinuities in the strand(s) of the DNA double helix. The can be single strand breaks (SSBs) where only one strand of the DNA molecule is affected or double strand breaks (DSBs) where both strands of the DNA are affected. |
| Transcription | The process by which the information in a strand of DNA is copied into a new molecule of messenger RNA (mRNA). It is the first step in protein synthesis. |
| Transcription factor IIH (TFIIH) | In NER, TFIIH is a multiprotein complex, comprised of different proteins including XPB and XPD. |
| Transcription-coupled repair (TCR) | A subpathway of NER, it removes lesions from the template DNA strands of actively transcribed genes. |
| Xeroderma Pigmentosum (XP) | A genetic disorder in which there is a decreased ability to repair DNA damage caused by ultraviolet (UV) light due to defective NER. |
References
Borges, H. L., Linden, R., & Wang, J. Y. (2008). DNA damage-induced cell death: lessons from the central nervous system. Cell research, 18(1), 17–26.
Friedberg, E. C. (2008). A brief history of the DNA repair field. Cell research, 18(1), 3-7.
Kuzminov, A. (2001). Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proceedings of the National Academy of Sciences, 98(15), 8241-8246.
Lucero, R., & Horowitz, D. (2019). Xeroderma Pigmentosum. In StatPearls [Internet]. StatPearls Publishing.
Mehta, A., & Haber, J. E. (2014). Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harbor perspectives in biology, 6(9), a016428.
Prasad, R., Dyrkheeva, N., Williams, J., & Wilson, S. H. (2015). Mammalian base excision repair: functional partnership between PARP-1 and APE1 in AP-site repair. PloS one, 10(5), e0124269.
Schärer O. D. (2013). Nucleotide excision repair in eukaryotes. Cold Spring Harbor perspectives in biology, 5(10), a012609.