SCIENTIFIC COMMUNIQUÉ IX
Beyond Pigment, the Melanocortin 1 Receptor (MC1R) in DNA Repair
March 2021
Abstract
SCIENTIFIC COMMUNIQUÉ VIII explores how our skin cells instigate DNA damage responses (DDRs) following the induction of DNA damage by ultraviolet (UV) and high energy visible (HEV) light. We are exposed to profuse amounts of UV and HEV light daily from our environment and other artificial sources.
Our skin’s DDRs coordinate cell cycle transitions, DNA repair and cell termination, activities that work to coordinate the removal of carcinogenic DNA damage from our skin’s DNA. Melanocortin 1 receptor (MC1R) stimulation by alpha-melanocyte stimulating hormone (α-MSH) is now understood to enhance these DDR pathways, helping ensure irradiation derived DNA damage or lesions (the precursor of cancerous mutations) are removed from our skin cells quickly so that the overall health of our skin is maintained and, more importantly, life threatening skin cancer is prevented.
Afamelanotide is a potent analogue (equivalent) of α-MSH. It not only mimics α-MSH’s physiological actions but can do so more effectively than the natural hormone. This leads to the clinical hypothesis that afamelanotide has the potential to further increase our skin’s DNA repair capacity against UV and HEV light damage. Its clinical application should be relevant to populations that are at a greater risk of chronic photodamage – and hence the development of skin cancers – by supporting our native DDRs, therefore preventing the survival of cutaneous cancer cells.
One Gene, Many Variations
The “wild type” (non-variant or normal) MC1R gene is found predominantly expressed in individuals originating from regions that lie close to the equator. In these regions, inherent photoprotective qualities are essential to preventing sun damage and skin cancer, ultimately ensuring survival. The MC1R is involved in the production of eumelanin (brown/black pigment) within the skin and hair. Eumelanin acts as a natural barrier against UV light, absorbing some UV radiation before it is able to infiltrate our skin cells and cause any damage.
More than 50,000 years ago, large populations of humans migrated away from East Africa and into regions that lay outside of the tropics, where solar environments were less extreme. Although unknown at the time, our skin requires some exposure to UV light (UVB especially) to make vitamin D, a steroid which is not easily sustained through dietary consumption only. With vitamin D required for bone and neuromuscular health, the high levels of eumelanin pigment in the skin suddenly became disadvantageous to the survival of populations who had settled in the northern hemisphere and were no longer exposed to the same intensity of UV radiation. Adequate UVB absorption was now no longer achievable which, in turn, lead to these migrants suffering from vitamin D deficiency.
The sequence of our DNA (or genes) changes over time, a completely random phenomenon that creates diversity in the human gene pool. Certain variations in the MC1R gene that caused a deviation away from normal eumelanin production were now incredibly beneficial, allowing ancestral migrants to thrive and survive long enough to reproduce, thereby passing this gene to their offspring. As a response to the pressures of a new environment, the frequency of these “loss of function” variants of the MC1R gene increased considerably in northern hemisphere migrants, and at a much greater pace than other comparable genes (including other members of the MCR subfamily).
The MC1R gene is highly polymorphic (“poly” meaning many and “morphic” meaning shape) signifying many forms of this single gene still exists in the human population. It is now estimated there may be more than 100 different variations of the MC1R gene which all encode for a slightly different structure of the same receptor (MC1R). But in modern society, the survival advantage that a “loss of function” variant once held is no longer as applicable. Depicted in SCIENTIFIC COMMUNIQUÉ IV, those who have inherited a reduced function variant of the MC1R gene have a heightened sensitivity to UV light, with the cells of their skin vulnerable to damage from the sun and other UV light sources. Consequently, these populations are genetically prone to prematurely developing skin cancer. The life expectancy just 500 years ago was less than half of what it is today. Historically, most did not live to an age where skin cancer had much influence on life (it was much more likely that unsanitary living conditions or starvation would inflict death before skin cancer would take its toll). Now the average global life expectancy for both sexes is around 73.2 years and the number of skin cancer diagnoses continues to grow. In the US alone, it is indicated that more than 20,000 people die of skin cancer every year.
More Than Just Pigment
Initially, the link between “loss of function” MC1R variants and increased rates of skin cancer was attributed to the decrease in the production of eumelanin pigments (melanogenesis, otherwise known as the tanning response). Whilst it is true that more eumelanin equates to increased UV absorption (thus preventing the irradiation of our skin cells), its protective role had been overestimated.
A study published in 2000 by Palmer et al identified individuals who would normally have been considered at a lower risk of skin cancer development due to their darker complexion were, in fact, at similar risk to their fellow fair skin study participants. These individuals were carriers of a MC1R “loss of function” variant that, whilst still retaining its sustained melanogenic function, did not ensue melanoma resistance. This observation suggested other functions of the MC1R that are independent of its well-known pigmentary effect. In the past decade, further research has demonstrated a secondary physiological effect following MC1R’s activation that precedes pigment production. In SCIENTIFIC COMMUNIQUÉ VIII DNA damage responses (DDRs) were introduced, specifically discussing our innate resistance to moderate UVR and HEV light damage. We now know the MC1R signalling axis confers a multitude of cutaneous photoprotective properties including that of DDR enhancement. They are:
1. Increased eumelanin synthesis (see SCIENTIFIC COMMUNIQUÉ I);
2. Antioxidative effects through melanogenesis and by increasing the expressions of antioxidative enzymes;
3. Elimination of photoproducts by enhancing the DNA repair pathway nucleotide excision repair (NER);
4. Enhancement of alternative cellular DNA repair systems: Base excision repair (BER) and Homologous Recombination (HR); and
5. Prevention of cell apoptosis (death)
It is estimated more than 30% of the world’s population carry a “loss of function” variant of the MC1R gene. This translates to more than 2 billion individuals at an even greater risk of skin cancer. Understanding MC1R variants, and thus being able to rescue some of their activity, has never been so prominent. In this communiqué, we will break down MC1R signalling in relation to DDRs and explain how, through pharmacological manipulation, there is the potential to reduce the rising incidence of skin cancer.
*This communique will build on the knowledge detailed in SCIENTIFIC COMMUNIQUÉ VI and SCIENTIFIC COMMUNIQUÉ VIII.
MC1R Signalling Axis and Genome Stability
After our skin is exposed to UV light from either the sun or artificial sources, our keratinocytes produce (synthesise) and release the signalling molecule α-MSH. This molecule binds to nearby MC1Rs found on the surface of various skin cells, including melanocytes, keratinocytes and fibroblasts. In doing so, α-MSH activates the receptor, first causing a conformational change which then activates what is known as the MC1R signalling axis. Part of the signalling axis is involved in melanogenesis in our melanocytes. In all our cutaneous cells, however, this signalling axis also enriches their DDRs (see SCIENTIFIC COMMUNIQUÉ VIII).
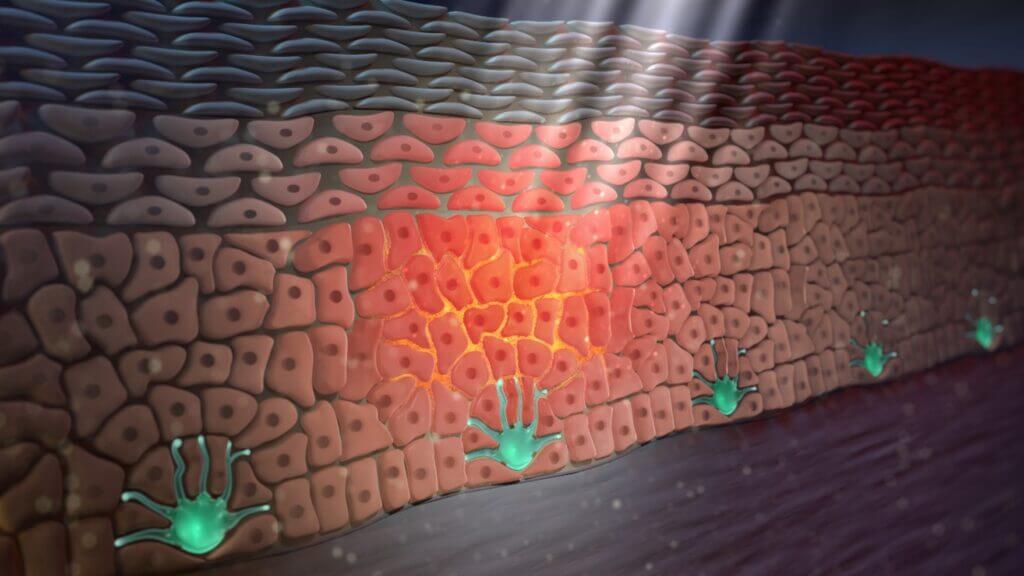
Figure 1: UVB light triggers the synthesis of α-MSH (orange) from the skin’s keratinocytes. α-MSH is then released where it binds to the MC1Rs on the surface of other skin cells, including the keratinocytes themselves.
Conformational Change of MC1R Following Activation and DNA Repair
In 2014, a research group at the University of Cincinnati, led by Prof Swope, were investigating the emerging theory that the MC1R was involved in augmenting our skin cells’ response to UVR damage. Based on earlier work by preceding scientists, the group hypothesised that the wild type (non-variant or “normal”) MC1R affected our skin’s DDRs and increased their UV and HEV light damage repair capacity. Attempting to characterise this notion further, the group exposed human melanocyte cell cultures to UV radiation, then incubated the cells with α-MSH, measuring the subsequent changes. They compared any differences observed to a control culture of MC1R absent melanocytes which were also exposed to the same conditions. This led to a fascinating discovery.
While both samples of cells immediately responded to UV exposure as expected (attempting to correct the damage caused), α-MSH’s stimulation of the MC1Rs helped enhance the concentration of certain DNA damage responding proteins. One of these proteins was xeroderma pigmentosum C (XPC). Swope never detailed why this rise in XPC was seen, but Ming et al (2012) had already depicted this, two years prior. Phosphatase and tensin homolog (PTEN), a phosphatase protein, normally exists in our skin cells in extremely low numbers. This is because it is constantly being degraded. When the MC1R is activated, however, the conformational change of the receptor allows PTEN to physically interact with it. This prevents PTEN’s destruction and instead this phosphatase can now provoke the synthesis of copious XPC proteins.
XPC is a DNA damage sensor protein, meaning it can differentiate between our normal DNA sequence and the presence of a DNA lesion. Specifically, XPC detects the large bulky photoproducts created by our DNA’s absorption of UVB radiation. This is the first step in the NER process and enables subsequent NER stages to take place, eventually removing the lesion and rejuvenating the cell back to a stable healthy state.
One needs to follow the pieces of the puzzle. It is generally known in the scientific community that UV light diminishes the concentration of XPC in our skin. It is, therefore, apparent that MC1R’s activation can ‘rescue’ the NER pathway by ensuring all photoproducts are recognised by the NER mechanism. The absence of XPC (and its counterpart xeroderma pigmentosum E, XPE) prohibits our cells from recognising potentially carcinogenic lesions in our DNA, thus eventually leading to cancerous cells escaping the repair process and developing into skin tumours. Interestingly, “loss of function” MC1R variants have a defect which reduces the receptor’s ability to interact with PTEN, inhibiting adequate repair of photolesions and greatly increasing an individual’s skin cancer risk. Ray et al (2016) explained a further function of XPC as part of the skin’s protective response to UV damage. XPC can increase the activation of two significant enzymes, namely ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and RAD3-related (ATR). Both fall into a class of proteins known as phosphatidylinositol-3 kinase-related kinases (PIKKs) which each function by modifying the activity of other molecules involved in our skin cell’s DDR network to favour DNA repair or cell termination.
While UV-induced DNA damage is sufficient to activate the PIKKs independent of XPC, the XPC protein further heightens their activation which leads to greater DDR efficiency. In SCIENTIFIC COMMUNIQUÉ VIII, checkpoint proteins were introduced as molecules which temporarily stall the cell in its current cell cycle phase to enable time for the repair to take place. The PIKKs are the enzymes which “switch on” these checkpoint proteins. This was also confirmed by Swope’s earlier experiments. The PIKKs also help convert a protein known as H2A histone family member X (H2AX) into γH2AX. γH2AX is required for recruitment of specific DNA repair proteins to DNA lesions, including XPC and XPE, in order to trigger NER, as well as other proteins involved in the repair of double-strand breaks (DSBs) which are also caused by UV and HEV light damage. Swope did not observe an additional enlargement of γH2AX by α-MSH in the non-functional MC1R melanocytes, consistent with their inability to respond to α-MSH with enhanced DNA repair.
In general terms, Swope’s results showed the MC1R signalling axis is urgently needed to help coordinate various aspects of our skin cells’ DDR including NER, HR and cell cycle transitions.
The MC1R Signalling Axis and DNA Repair
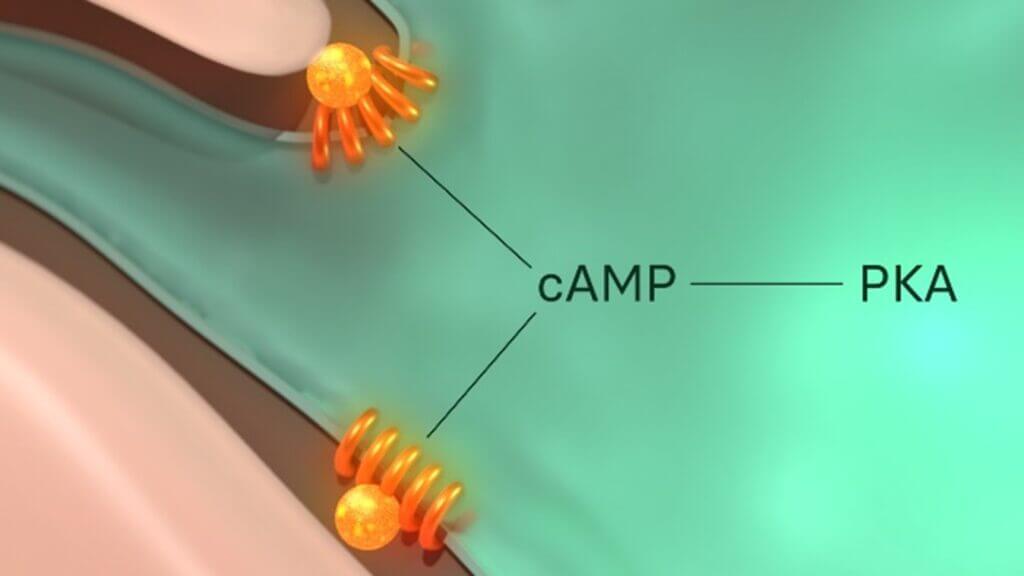
Figure 2: The MC1R is functionally coupled to adenylyl cyclase (not shown), a stimulator of cyclic adenosine monophosphate (cAMP). cAMP is a secondary messenger, meaning its role is to transduce the external signal dictated by α-MSH to numerous intracellular downstream effectors. cAMP therefore propagates and amplify α-MSH signal internally to cause its desired response from the cell.
Figure 2 illustrates the initial components of the MC1R signalling axis. While just the structural change of the receptor alone clearly influences a positive impact on our skin’s DDRs as mentioned above, its protective effects expand further. One immediate target of the MC1R signalling axis is protein kinase A (PKA). PKA is a kinase enzyme, which means it operates by chemically altering other molecules through a process known as phosphorylation. PKA has multiple targets, one of which is ATR. Although it was previously mentioned that the activation of ATR is induced by XPC, ATR is subsequently required to enable the actions of another xeroderma pigmentosum protein, xeroderma pigmentosum A (XPA), the damage verification protein in this repair system. This differs from XPC’s role as it appears much later in the NER pathway. It is the final confirmation to the cell that there is indeed a photolesion in its DNA that must be removed. This extra verification step prevents the accidental removal of a normal section of DNA that has been mistaken for damage, which would otherwise introduce an additional array of problems. XPA also helps assemble the incision proteins – proteins that cleave the lesion away from the rest of the otherwise healthy DNA during NER.This signifies the importance of PKA’s activation, through the MC1R signalling axis, to increase the efficacy of the DNA repair mechanisms NER and BER in skin cells damaged by UV and HEV light. Additionally, the same PKA molecules can act to reduce the level of DNA damage caused by ROS by increasing our natural antioxidant levels.
Unfortunately, XPA resides naturally in the cytoplasm of the cell, the area that is outside of the nucleus (where our DNA is kept). Only when XPA is bound to ATR can it be transported (translocated) into the skin cell’s nucleus where it localises to the area surrounding the lesion. This is because ATR can associate itself with importins that allow for the translocation process. By phosphorylating ATR, PKA is enhancing ATR’s binding capabilities to XPA and therefore boosting the NER process.
A second target of PKA is cAMP response element-binding protein (CREB). CREB obliquely increases the expression of transcription factor IIH (TFIIH). Reviewed in SCIENTIFIC COMMUNIQUÉ VIII, TFIIH is a multicomplex protein composed of several proteins, including xeroderma pigmentosum B (XPB) and xeroderma pigmentosum D (XPD). TFIIH helps unwind our DNA around the damage site to increase the access for other NER proteins and therefore enables the precise removal of the photolesion.
PKA’s actions are not limited to aiding the NER process. Through both direct and indirect means , PKA also activates the transcription factor p53. In its phosphorylated state, p53 can interact with the cell’s genome, switching on the expression of target genes and thereby controlling the production of specific proteins. Base excision repair (BER) is the DNA repair pathway that restores the DNA damage caused indirectly by UVA and HEV light. Both these wavelengths generate a build-up of reactive oxygen species (ROS) within our skin cells, which react and oxidise our DNA (see SCIENTIFIC COMMUNIQUÉ VI). Two enzymes are key to the BER process: 8-oxoguanine DNA glycosylase 1 (OGG1) and apurinic/apyrimidinic endonuclease 1 (APE1). Both facilitate the removal of oxidised lesions. Importantly, adequate and precise activation of the MC1R signalling axis leads to the transcriptional upregulation of both OGG1 and APE1 by p53. This increase in protein expression has been linked with an increase in cellular resistance to radiation as shown in a study completed by Kadekaro el at (2012) p53’s protective role against DNA oxidation is not limited to its influence modulating the BER pathway, but concomitantly encourages the synthesis of antioxidants such as catalase and ferritin. These molecules function to “soak up” surplus ROS, thereby reducing the number of oxidised lesions needed to be repaired in the first place. Barckhausen et al (2013) suggested p53 may also be a regulator of XPC, expression but this requires further clarification.
MC1R Signalling and Cell Survival
One in three Americans have admitted experiencing at least one sunburn in the past year. Despite sunburn being recognised as an acute injury, incurring multiple sunburns is a strong predictor of future skin cancer, particularly melanoma, the deadliest form of skin cancer. Sunburn appears as reddened inflamed skin which is warm to the touch and commonly associated with mild to moderate pain. At a histological (cellular) level, our keratinocytes are undergoing cell death (apoptosis). They have been exposed to lethal doses of UV light and are no longer candidates for various repair processes as the level of DNA damage acquired exceeds the body’s DNA repair capacity. In an attempt to restrict the replication of DNA damage, these cells self-activate their destruction in an efficient and orderly manner. This is a successful protective measure against the carcinogenic effects of UVR: if these mutant cells were allowed to continue residing in the skin they would continue to grow and proliferate, eventually forming tumours.
Our melanocytes, on the other hand, are much more resistant to radiation-induced apoptosis. Melanocytes have a very long turnover time, around three years. Unlike our plentiful keratinocytes, melanocytes make up only a fraction of our skin’s total cell count. If our melanocytes underwent cell death every time we experience a sunburn, our bodies would eventually run out of melanocytes and we would be unable to produce the much-needed protective eumelanin pigment. Akin to albinism, we would start experiencing skin cancer onset much earlier in life. So instead, when these cells are exposed to high levels of radiation damage, they survive. This has some extreme drawbacks. If they accumulate too much damage from repetitive sun damage that cannot all be repaired, some lesions remain ingrained in the DNA of melanocytes, eventually leading to melanoma.
Coa et al (2013) showed that MC1R regulates a survival pathway known as PI3K/Akt. This pathway does not typically threaten malignant transformation, and is actually required for the normal survival of a cell. Yet, when PI3K/Akt dominates in a cell containing unrepaired DNA damage, it increases the cell’s likelihood of propagating before it can repair or self-destruct. The PI3K/Akt pathway has been shown to increase angiogenesis, a process by which new blood vessels grow from existing ones. Tumour progression relies on a continuous supply of oxygen and nutrients, with angiogenesis enabling the development of blood vessels which consequently penetrate the cancerous cells. This allows for not only tumour growth and survival but also metastasis (cancer spreading) as the blood supply is utilised as a route for cancer migration. PTEN is an inhibitor of the PI3K/Akt survival pathway, moderating its activity. A reduction of PTEN is also a “signature” in melanoma cells. While MC1R’s protection of PTEN degradation can therefore help mitigate melanoma risk, “loss of function” MC1R variants lack this capacity, helping explain why these populations are at a high risk of melanoma.
Finally, the MC1R signalling axis also has a mitogenic effect on both these keratinocytes and melanocytes. This is because of PKA’s upregulation of peroxisome proliferator-activated receptor gamma (PPAR-γ). In the likelihood a DNA lesion has escaped the cell’s DDRs, as a final measure to prevent tumour transformation, the PPAR-γ signalling pathway function to inhibit excessive cell proliferation.
Manipulating α-MSH as the Enhancer
It is important to note that α-MSH does not inaugurate DNA repair for the most part, but rather enhances its effects. Our skin is the primary barrier to external threats, including that of UV and HEV light, and skin cells protect our internal tissues from environmental damage by bearing most of the trauma. Thus, our skin cells must be extremely robust and capable of regeneration, with amplified DNA repair a substantial part of this trait. But mutations mean some individuals’ health would benefit greatly from enhanced processes.
Those who have inherited a “loss of function” MC1R variant are generally still able to exert DDRs following DNA injury. Yet their intrinsic DDRs are operating at a reduced competence, meaning they are unable to handle the continuous and long-term effects of sun damage and placing them at much higher risk of skin cancer. Skin cancer recurrence is also extremely likely in these individuals, with 60% of those who developed a skin cancer being diagnosed with a second malignancy within 10 years. Even when it can be removed, skin cancer surgery is extremely abrasive, leaving many with visible scars. In the case of malignancy, aggressive and more invasive treatment is needed which comes with very serious side effects.
One solution has been to attempt to re-optimise the MC1R signalling axis through pharmacological manipulation. And yes, this is the core knowledge and business of CLINUVEL. The “loss of function” MC1R variants are, however, less effectually activated by α-MSH. Studies by Minder et al (2017) and Fitzgerald et al (2005) identified that the synthetic analogue afamelanotide successfully rescues receptor functionality. Targeting the MC1R directly, as opposed to the receptor’s downstream effectors, lessens off-target effects that might otherwise introduce complications in the form of an adverse reaction or unintended effect. This also provides a therapeutic option for other populations whose repair capacity has been compromised by other means outside the MC1R variants. This includes organ transplant recipients and xeroderma pigmentosum (XP) patients.
Conclusion
The MC1R has a multitude of roles systematically, and we have an increasing understanding of MC1R as a major regulator of skin biology foregoing melanogenesis. Exploiting the cutaneous melanocortin system, therefore, has the potential to act as a preventive strategy towards skin cancer, which carries significance in individuals prone to this life-threatening disease. Further research in human trials is, however, needed to clinically confirm this proposition.
Useful Links
This SCIENTIFIC COMMUNIQUÉ IX is the final in a series of three COMMUNIQUÉs on cutaneous DNA damage and repair in humans. The reader is advised to read all three COMMUNIQUÉs collectively. Links to the preceding COMMUNIQUÉs can be found here:
SCIENTIFIC COMMUNIQUÉ VI – Ultraviolet Radiation Damage and Oxidative Stress in Skin Cancer
SCIENTIFIC COMMUNIQUÉ VIII – DNA Repair Mechanisms
Download PDF
Glossary
| Term | Definition |
| 6-4 PP | Pyrimidine 6-4 pyrimidone photoproducts. A DNA mutation formed as a result of UVB radiation exposure. |
| Adenylyl cyclase (AC) | Membrane bound enzyme that converts the formation of cyclic AMP from ATP. It is known as a primary messenger and can be found in numerous intracellular signalling pathways. |
| Akt | V-akt murine thymoma viral oncogene homolog (Akt), a serine/threonine-specific protein kinase. |
| α-Melanocyte-Stimulating Hormone | Also known as alpha-MSH or α-MSH. A natural hormone and neuropeptide that is part of the melanocortin system. |
| Apoptosis | A form of programmed cell death. |
| Ataxia Telangiectasia Mutated (ATM) | A protein kinase usually activated by double stranded DNA breaks. If the breaks are not repaired ATM initiates a signalling cascade that cumulates in cell cycle arrest. |
| ATM and RAD3-related (ATR) | A protein kinase activates by DNA damage. If damage remains unrepaired ATR initiates a signalling cascade that cumulates in cell cycle arrest. |
| Base Excision Repair (BER) | DNA repair pathway in which a single faulty base is removed from the DNA and replaced. |
| Cancer | Disease featuring abnormal and improper controlled cell division resulting in invasive growths, or tumours, that may spread throughout the body. |
| Cell Division | Separation of a cell into two daughter cells. |
| CREB | cAMP response element binding protein. |
| Cyclic Adenosine 3,5 -Monophosphate (cyclic AMP or cAMP) | A nucleotide that is generated from ATP by adenylyl cyclase in response to various extracellular signals. It is a small signalling molecule that activates cAMP dependent proteins such as protein kinase A. |
| Cyclobutane Pyrimidine Dimers (CPDs) | Molecular lesions formed from thymine or cytosine bases in DNA via photochemical reactions. |
| Gamma H2A Histone Family Member X (γH2AX) | The phosphorylated form of H2AX. |
| H2A Histone Family Member X (H2AX) | A type of histone protein. |
| Histone | Small proteins that form the nucleosome core around which DNA is wrapped around in eukaryotic chromosomes. |
| Homologous Recombination (HR) | Genetic exchange between a pair of identical or very similar DNA sequences, typically those located on two copies of the same chromosome. |
| Keratinocytes | The primary cell type found in the epidermis. |
| Malignant | Invasive tumours that are able to undergo metastasis. A malignant tumour is a cancer. |
| Melanocortin 1 Receptor (MC1R) | A transmembrane receptor that is expressed by a variety of cells including the skin cells. It is part of the melanocortin system. |
| Melanocyte | A cell that produces melanin. |
| Melanoma | A malignancy originating from the melanocyte and now known to be linked to a variety of biochemical and genetic defects. Melanoma is an umbrella term for a variety of tumours with diverse biological behaviour. |
| Mutation | Heritable change in the nucleotide sequence of the DNA. |
| Nucleotide Excision Repair (NER) | Type of DNA repair that corrects damage of the DNA double helix, such as those caused by UV damage, by removing the damage region on one strand and resynthesising it using the undamaged strand as a template. |
| Oxidation | Loss of electrons from an atom. |
| p53 | Tumour suppressor gene found mutated in 50% of human cancers. Encodes a gene regulatory protein (transcription factor_ that is activated by damage to DNA and is involved in blocking further progression through the cell cycle. |
| Peroxisome Proliferator Activated Receptor Gamma (PPAR-γ) | A nuclear receptor. |
| Phosphatase and Tensin Homolog (PTEN) | A phosphatase protein. It plays a critical role in cell growth, proliferation and differentiation. |
| Phosphatidylinositol-3-kinase (PI3K) | A membrane bound enzyme that is a component of the PI3K/Akt signalling pathway. |
| Protein kinase A (PKA) | An enzyme that phosphorylates target proteins in response to a rise in intracellular cAMP levels. |
| Reactive Oxygen Species (ROS) | A reactive chemical species containing oxygen, such as the superoxide anion or hydrogen peroxide. |
| Transcription Factor IIH (TFIIH) | In NER, it functions as a helicase that unwinds DNA. |
| Transcription Factor | Term loosely applied to any protein required to initiate or regulate transcription in eukaryotes. Includes gene regulatory proteins, the general transcription factors, co-activators, co-repressors etc. |
| Tumour | Abnormal mass of cells resulting from a defect in cell proliferation control. A tumour can be non-invasive (benign) or invasive (cancerous, malignant). |
| Xeroderma Pigmentosum (XP) | A genetic disorder in which there is a decreased ability to repair DNA damage caused by ultraviolet (UV) light due to defective NER. |
| XPA | Xeroderma pigmentosum, complementation group A, the photo-mutation verification protein which acts as a scaffold protein for XPG and XPF during NER. |
| XPB | Xeroderma pigmentosum, complementation group B, a DNA helicase, unwinding the DNA from the 5’ to 3’ end during NER. Critical component of the TFIIH complex. |
| XPC | Xeroderma pigmentosum, complementation group C, the DNA lesion identification protein in global genome NER facilitating the recruitment of the TFIIH complex to the site of the photomutation. |
| XPD | Xeroderma pigmentosum, complementation group D, a DNA helicase, unwinds the DNA from the 3’ to 5’ end during NER. Critical component of the TFIIH complex. |
| XPE | Xeroderma pigmentosum, complementation group E, a damage binding protein in the NER process. |
| XPF | Xeroderma pigmentosum, complementation group F, an endonuclease which cleaves a single strand of DNA on 5’ side of the photomutation during NER. |
| XPG | Xeroderma pigmentosum, complementation group G, an endonuclease which cleaves a single strand of DNA on 5’ side of the photomutation during NER. |
References
Barckhausen, C., Roos, W. P., Naumann, S. C., & Kaina, B. (2014). Malignant melanoma cells acquire resistance to DNA interstrand cross-linking chemotherapeutics by p53-triggered upregulation of DDB2/XPC-mediated DNA repair. Oncogene, 33(15), 1964-1974.
Cao, J., Wan, L., Hacker, E., Dai, X., Lenna, S., Jimenez-Cervantes, C., Wang, Y., Leslie, N. R., Xu, G. X., Widlund, H. R., Ryu, B., Alani, R. M., Dutton-Regester, K., Goding, C. R., Hayward, N. K., Wei, W., & Cui, R. (2013). MC1R is a potent regulator of PTEN after UV exposure in melanocytes. Molecular cell, 51(4), 409–422.
Fitzgerald, L. M., Fryer, J. L., Dwyer, T. & Humphrey, S. M. Effect of MELANOTAN1, [Nle4, D-Phe7]-a-MSH, on melanin synthesis in humans with MC1R variant alleles. (2005).
Flori, E., Rosati, E., Cardinali, G., Kovacs, D., Bellei, B., Picardo, M., & Maresca, V. (2017). The α-melanocyte stimulating hormone/peroxisome proliferator activated receptor-γ pathway down-regulates proliferation in melanoma cell lines. Journal of experimental & clinical cancer research: CR, 36(1), 142.
Kadekaro AL, Chen J, Yang J, Chen S, Jameson J, Swope VB, Cheng T, Kadakia M, Abdel-Malek Z. Alpha-melanocyte-stimulating hormone suppresses oxidative stress through a p53-mediated signaling pathway in human melanocytes. Mol Cancer Res. 2012 Jun;10(6):778-86.
Karar, J., & Maity, A. (2011). PI3K/AKT/mTOR pathway in angiogenesis. Frontiers in molecular neuroscience, 4, 51.
Minder, E. I., Barman-Aksoezen, J. & Schneider-Yin, X. Pharmacokinetics and Pharmacodynamics of Afamelanotide and its Clinical Use in Treating Dermatologic Disorders. Clin Pharmacokinet 56, 815–823 (2017).
Ming, M., & He, Y. Y. (2012). PTEN in DNA damage repair. Cancer letters, 319(2), 125–129.
Palmer JS, Duffy DL, Box NF, Aitken JF, O’Gorman LE, Green AC et al. (2000) Melanocortin-1 receptor polymorphisms and risk of melanoma: is the association explained solely by pigmentation phenotype? Am J HumGenet 66:176–86
Ray, A., Blevins, C., Wani, G., & Wani, A. A. (2016). ATR- and ATM-Mediated DNA Damage Response Is Dependent on Excision Repair Assembly during G1 but Not in S Phase of Cell Cycle. PloS one, 11(7), e0159344.
Swope, V., Alexander, C., Starner, R., Schwemberger, S., Babcock, G., & Abdel‐Malek, Z. A. (2014). Significance of the melanocortin 1 receptor in the DNA damage response of human melanocytes to ultraviolet radiation. Pigment cell & melanoma research, 27(4), 601-610.
The Skin Cancer Foundation. 2020. Skin Cancer Facts & Statistics – The Skin Cancer Foundation.