SCIENTIFIC COMMUNIQUÉ VI
Ultraviolet Radiation Damage and Oxidative Stress in Skin Cancer.
October 2020
Introduction
Despite government campaigns to curb increases, current statistics show the rate of skin cancer is escalating, particularly in regions with predominantly fair-skinned populations. Skin cancer diagnoses have increased considerably over the last five decades, at a rate of 4% per annum. Approximately 5.3 million individuals are diagnosed with a skin cancer in the USA annually. Two in three Australians will be diagnosed with a form of skin cancer by the age of 70. Advances in molecular genetics in recent decades have led to a better understanding of the aetiology of skin cancers, which must now be used to help combat the rise of these life-threatening conditions.
Skin cancers are an abnormal growth of cells within skin tissue. As discussed in SCIENTIFIC COMMUNIQUÉ IV there are three main types of skin cancer: basal cell carcinoma (BCC), squamous cell carcinoma (SCC) and various forms of melanoma. Common to all three is a pathogenesis which is underpinned by overexposure of skin to genotoxic agents. Ultraviolet radiation (UVR) is the major source, with increasing evidence showing that high energy visible (HEV) light is also a contributor.
Both UVR and HEV light damage skin integrity, but their effect on cellular DNA is arguably the most detrimental: initiating DNA specific chemical alterations, which leave cells unstable – the preliminary step in cancer formation. The human body has evolved DNA repair systems that identify and restore these impairments, but the capacity of these DNA repair mechanisms is limited, with excessive DNA damage associated with inefficient restoration. In such cases, the probability of skin cancer rises markedly. With roughly 90% of the world’s population residing in regions where the peak annual UV index reaches 10 (out of a possible 11-point scale), this risk is exacerbated by the innate threat of solar induced damage to the skin. Furthermore, genetic predispositions in individuals with lighter skin types naturally increases their susceptibility to skin cancer formation (see SCIENTIFIC COMMUNIQUE IV). However, while certain genetic influences and disorders can reduce an individual’s DNA repair capacity, it is generally recognised that unsafe behaviour towards sun exposure is the most prevalent factor in the high rates of skin cancer seen within the general population.
Understanding the mechanisms behind solar-radiation-induced DNA damage is a crucial step in reducing the rising incidence of skin cancer. In this SCIENTIFIC COMMUNIQUÉ, we will review the clinical significance of skin cancer as a disease and then look closely at the aetiology behind its formation. Successive COMMUNIQUÉs will then examine endogenous DNA repair mechanisms and review the use of potential innovative therapeutics to reduce the risk of these cancers.
Ultraviolet Radiation, DNA Damage and Skin Cancer
The skin has a multifaceted biological function, including the maintenance of systematic homeostasis through aiding the regulation of internal thermal temperatures and preventing water loss. Most importantly, the skin operates as a physical barrier, acting as the body’s first line of defence against exogenous agents. Whilst beneficial, this means the cells which compose the many layers of the skin are subjected to repeated environmental insults. This includes solar radiation, electromagnetic energy from the sun comprising three distinct wavelengths: UVR (200-400nm), visible light (400-700 nm, including HEV light at 400-500nm), and infrared (>700nm).
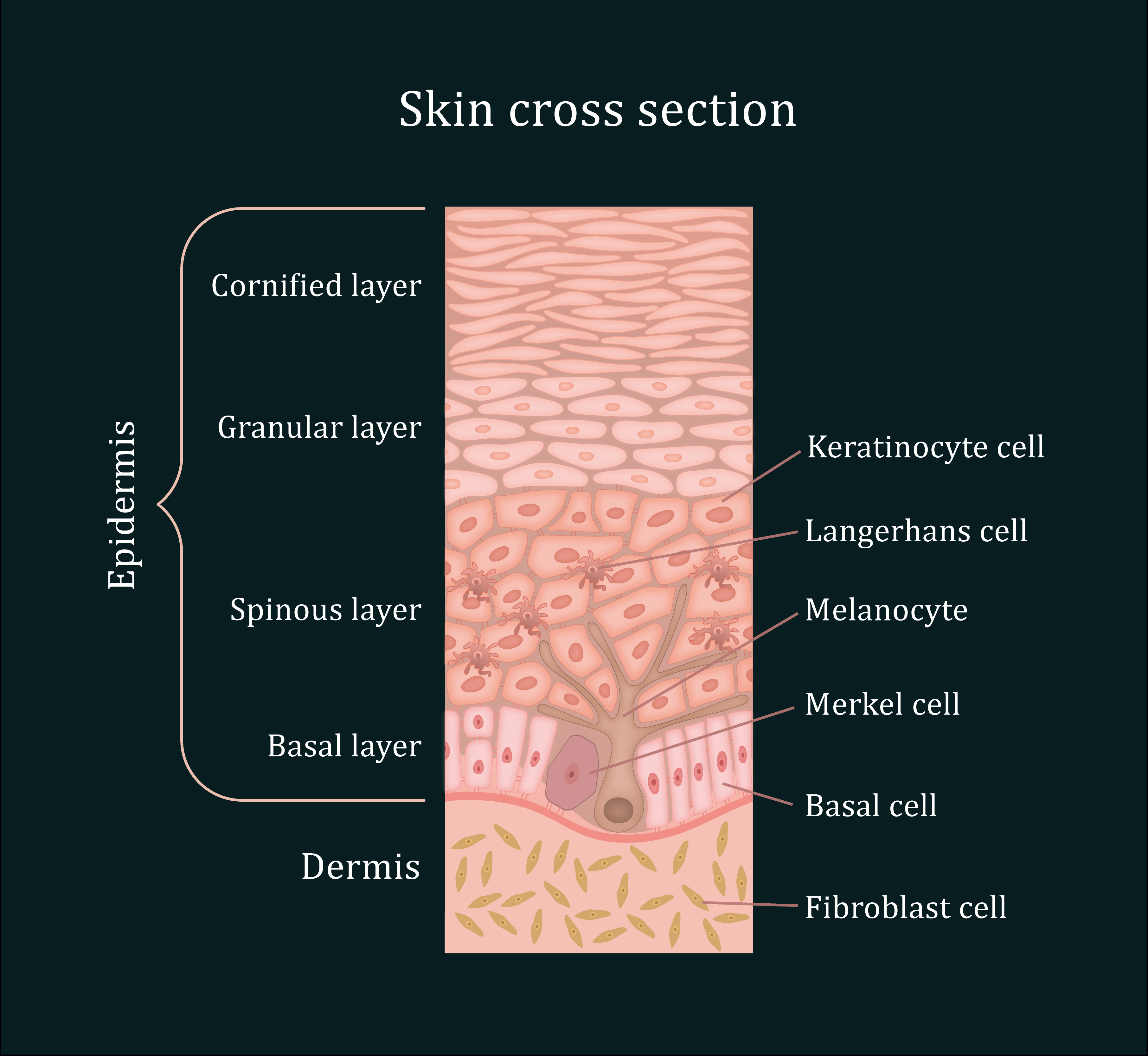
Figure 1: The organisation of the human skin. The skin is composed of three main layers: The epidermis, the dermis and the hypodermis (not shown). The epidermis is 4 to 5 cell layers thick, made of closely packed epithelial cells. 90% of the epidermis is composed of keratinocytes. The basal cells and melanocytes are found exclusively within the basal layer of the epidermis, with solar-induced damage initiating the formation of basal cell carcinoma (BCC) and melanoma, respectively, in these cells. Situated in the epidermal layer above lies the squamous keratinocytes. Similar to BCC and melanoma, squamous cell carcinoma (SCC) arises after an accumulation of pro-oncogenic mutations within the DNA of these cells.
Solar UVR and HEV light penetrates human skin, where they reach and react with DNA in the nucleus of various skin cells. These interactions damage the integrity of DNA leading to the formation of mutations. If left unrepaired, these mutations become permanently ingrained in the cell’s DNA. While not all mutations are immediately threatening, the origin of all skin cancers is found in the initiation of a permanent mutation which has been introduced into the genetic sequence of an oncogene. Such alterations cause the cell to break the normal restraints on cell proliferation, providing the mutated cell with growth and survival advantages over existing healthy cells. Rapid division and resistance to apoptosis (cell death) allows this cell and its subsequent generations to develop abnormally, known as dysplasia. While at this stage the cells remain precancerous, additional mutations confer further proliferative capabilities and promote local cell migration, leading to malignancy.
Skin cancer disturbs the skin’s normal function, with dermatological abnormalities arising as an initial symptom. The developing malignant tumours invade local cutaneous tissue corrupting the skin by disrupting cellular homeostasis. As a result, the skin becomes defective, leading to rapidly declining health as the body’s barrier against pathogenic threats and external stress is compromised. Skin cancers can also damage the underlying muscle, cartilage, and bone as they progress. Even with early intervention, these malignancies are known to cause permanent disfigurement.
The main skin cancers all arise from the atypical proliferation and growth of cells found in the epidermis (Figure 1). They can be readily distinguished by their visual presentation, behaviour, and metastatic capabilities (ability to spread to secondary sites). While all three cancers result from the accumulation of mutations within the cell’s proto-oncogenes, there exists some biopathological variation between the mutagenic profile of each cutaneous tumour which contributes to its pathogenesis.
The non-melanoma skin cancers, BCCs and SCCs, are characterised by slow growth and are more often pink in colour (Figure 2). Both BCCs and SCCs display a limited potential for metastasis, 0.55% and 5% respectively. BCCs are the most common skin malignancy in lighter skinned populations, accounting for an estimated 75% of all reported cases. They arises from mutations in the basal cells, which are involved in the production of new keratinocytes to replenish the loss of surface cells from the stratum corneum (Figure 1). Distinct from SCCs and melanomas, the development of BCCs occurs ex novo, meaning they do not arise from pre-cancerous lesions. Analysis of BCC tumours has shown solar radiation damage leads to the mutations of genes such as PTCH, which results in aberrant activity of the sonic hedgehog pathway. This cellular pathway is critical in mediating the self-renewal cell cycle of the basal cells. When lost, tumour formation is instigated.
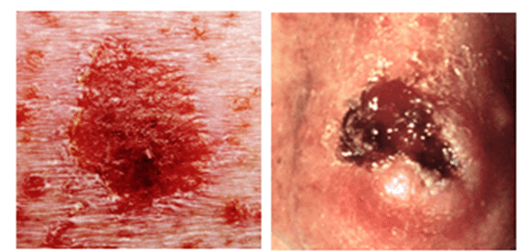
Figure 2: BCC (L) and SCC (R) visual characteristics BCC: Generally, a bump or skin growth, or it could be flat or slightly raised. Colours may be white or light pink; flesh-coloured or brown; or pearly, or waxy. BBC almost always arises ex novo. SCC: May present as firm, flesh-coloured, red or brown scaly papule or plaque, however they may also present as a non-healing ulcer, smooth nodule, conical horn or wart like growth. SCC is preceded by in situ or pre-lesion known as an actinic keratosis.
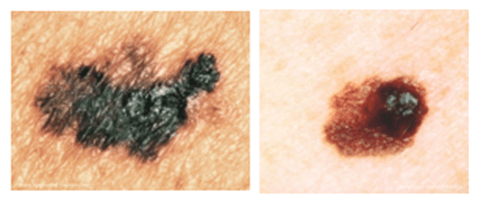
Figure 3: Melanomas may appear brown/black or pinkish in colour and irregular in shape as a result of the abnormal melanocytes retaining their ability to synthesis melanin pigment. However, reports of white melanomas are not unheard of. Their clinical appearance also shares similarities with harmless cutaneous moles which has known to contribute to late diagnosis and thus their undetected developmental progression as in up to 30% of cases of melanoma arise from existing pigmentary lesions.
Squamous cell carcinomas arise from the oncogenesis of keratinocytes that reside in the squamous cell layer of the epidermis. Actinic keratoses (AK) are the known pre-lesions of SCCs; roughly 65% of all SCCs develop from further progression of AK lesions. Mutations more frequently identified in SCCs include mutations in the NOTCH and NF-KΒ genes. As they are more likely to metastasise during the later stages of their development, SCCs account for 75% of all nonmelanoma skin cancer deaths.
A melanoma is a growth of dysregulated melanocytes and can be darkly pigmented (Figure 3). Though less common, accounting for 1% of all skin cancers in the USA, melanomas are much more aggressive and metastasise more rapidly, leading to a poor prognosis for patients. This increase in metastasising behaviour could be due to the upregulation of the mitogen activated protein kinase (MAPK) signalling pathway in these anomalous melanocytes. However, as in BBCs and SCCs, a complex mechanism of various pro-carcinogenic mutations is the more plausible contributor.
This highlights the determinantal effects of oncogenic mutations in a range of skin cells, with metastasis noted as the most severe consequence of overexposure to UVR and HEV light, ultimately leading to premature mortality if left untreated.
Photolesions: Direct DNA Damage by UVB
DNA damage can be a result of direct and indirect damage. UVA (320-400 nm) and HEV light (400-500 nm), are known to disrupt DNA through secondary mechanisms, while UVB (280-320 nm) causes direct damage. The shorter wavelength, UVB , corresponds to a more energetic (therefore more damaging) form of radiation. While the wavelength limits the ability of UVB to penetrate to the uppermost papillary layer of the dermis, its higher frequency contributes to more intense signs of cellular damage, including its ability to directly react with the DNA within the resident epidermal cells.
When skin is exposed to UVB radiation, the DNA within skin cells directly absorbs photon energy, resulting in the formation of two major classes of DNA mutations, cyclobutane pyrimidine dimers (CPDs) and pyrimidine 6-4 pyrimidone photoproducts (6-4PPs), at a ratio of 4:1.
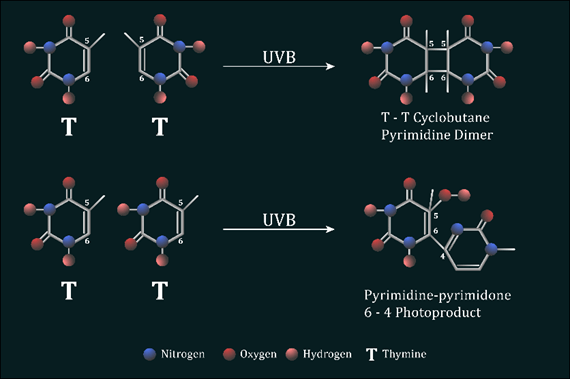
Figure 4: UVB induced photolesions The absorption of UVB energy results in the formation of a four-member ring structure which occurs as two abnormal covalent bonds are generated between carbons 5 and 6 of adjacent thymine or cytosine bases on the same DNA strand. A single non-cyclic covalent bond is created in the DNA, after reacting with UVB, between carbon 6 and 4 of neighbouring pyrimidines nucleotides on the same DNA strand to generate 6-4PPs
CPDs result from the addition of abnormal covalent bonds between carbons 5 and 6 of adjacent thymine or cytosine nucleotide bases. In contrast, a non-cyclic bond created between carbon 6 and 4 of neighbouring pyrimidines nucleotides generate 6-4PPs (Figure 4). Regardless of these disparities, the presence of an intra-strand cross-linking bond between dipyrimidine sites results in a large bulky distortion of the DNAs structure. These distortions have several effects. During cell division, the DNA replication complex, otherwise known as DNA polymerase, is stalled at the site of the lesion during fork progression as DNA replication occurs. This results in the collapsing of the DNA forks and thus the formation of double strand breaks. Cells containing double strand breaks must either undergo homologous or non-homologous recombination to resolve these breaks. Double strand breaks are considered the most dangerous DNA lesion as they cause chromosome discontinuity if left unrepaired, which ultimately leads to cell death. Alternatively, the abnormal linkage of two pyrimidine nucleotides within the DNA can often be misinterpreted by the DNA replication machinery as a singular nucleotide base. This means that, after normal cell division, there will be a nucleotide missing from the replicated DNA molecule found within the progeny cell, causing what is known as a frame shift mutation, corrupting the coding sequences of genes downstream of this change. This disruption has the potential to affect multiple gene sequences as the reading frame after the point of mutation is distorted, resulting in the production of numerous aberrant proteins which contribute to tumour transformation.
Reactive oxygen species (ROS)
Both UVR and HEV light react with photoreactive molecules found within the skin cells, such as riboflavin and porphyrin, generating large quantities of intracellular reactive oxygen species (ROS). ROS are a group of chemically reactive species, which also play a role in generating DNA mutations. All ROS molecules share the characteristic of containing one unpaired electron, thus they are very unstable and react rapidly with alternative molecules in the surrounding environment. There are a number of ROS examples found within the human body including hydrogen peroxide (H2O2), super oxide anions (O⁻ ₂) and singlet oxygen molecules (O=O), however arguably the most reactive and damaging ROS are the hydroxyl radicals. They are typically formed during the Fenton reaction and have minuscule diffusion distances (nanometres). Hydroxyl radicals actively target the DNA nucleotide guanine due to its low redox potential.
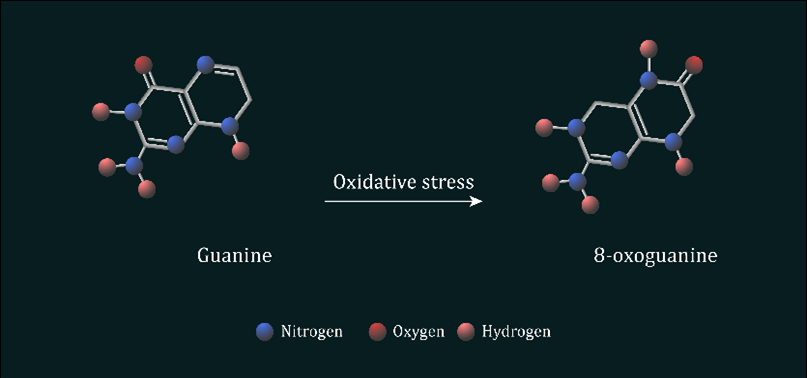
Figure 5: Oxidation of guanine nucleotide to form 8-oxo-7,8-dihydroguanine ROS such as hydroxyl radicals reacts with guanine nucleotides found within the DNA, which leads to the protonation of guanine’s alone pair of electrons at N7.
ROS mediated oxidation of guanine to 8-oxo-7,8-dihydroguanine (8-oxoG) alters the base’s chemical properties, allowing the now oxidated base to mimic the characteristics of a thymine nucleotide. During DNA replication atypical base pairing, known as Hoogsten pairing, is promoted and the oxidated guanine now pairs with adenine instead of cytosine. This process is known as transversion, as C:G is rapidly transitioned to A:T on the newly synthesised DNA strand. It is estimated up to 2,000 8-oxoG mutations form in mammalian DNA within a normal 24h period, with this figure only further increasing after excessive UVR and HEV light exposure.
Summary
The combination of photoproduct production alongside DNA oxidation, all instigated by solar radiation, leads to the accumulation of hundreds and thousands of mutations after overexposure to UVR and HEV light. With a singular mutation possessing the ability to disrupt normal cell function, the inability to repair such damage results in uncontrolled tumour formation. As a normal response against solar radiation insult, cellular DNA repair mechanisms are activated and upregulated in these cutaneous cells which work to remove potentially lethal mutations. In general, these mechanism work to uphold the skin’s integrity, albeit with some limitations to their functionality. The following SCIENTIFIC COMMUNIQUÉs will discuss these processes in greater depth and how they may be targeted to reduce the risk of skin cancers.
Useful Links
This SCIENTIFIC COMMUNIQUÉ IX is the first in a series of three COMMUNIQUÉs on cutaneous DNA damage and repair in humans. The reader is advised to read all three COMMUNIQUÉs in one breath. Links to the following COMMUNIQUÉs in the series can be found here:
SCIENTIFIC COMMUNIQUÉ VIII – DNA Repair Mechanisms
SCIENTIFIC COMMUNIQUÉ IX – Beyond Pigment, the Melanocortin 1 Receptor (MC1R) in DNA Repair
Download PdfGlossary
|
Term |
Definition |
|
6-4 PP |
Pyrimidine 6-4 pyrimidone photoproducts. A DNA mutation formed as a result of UVB radiation exposure. |
|
Apoptosis |
A form of programmed cell death. |
|
Basal Cell |
Found in the basal layer of the epidermis, they constantly divide to form new cells that have sloughed off the skin’s surface. |
|
Cancer |
Disease featuring abnormal and improper controlled cell division resulting in invasive growths, or tumours, that may spread throughout the body. |
|
Cyclobutane Pyrimidine Dimers (CPDs) |
Molecular lesions formed from thymine or cytosine bases in DNA via photochemical reactions. |
|
DNA Polymerase |
An enzyme that synthesises DNA by joining nucleotides together using a DNA template as a guide. |
|
Dysplasia |
The presence of abnormal cells within a tissue or organ. |
|
Fibroblast |
Common cell type found in connective tissue, for example the dermis of the skin. Secretes an extracellular matrix rich in collagen and other extracellular matrix macromolecules. Migrates and proliferates readily in wounded tissue and tissue culture. |
|
Keratinocytes |
The primary cell type found in the epidermis. |
|
Langerhans Cell |
Cutaneous-resident macrophages found in the epidermis layer of the skin. |
|
Malignant |
Invasive tumours that are able to undergo metastasis. A malignant tumour is a cancer. |
|
Melanocyte |
A cell that produces melanin (pigment). |
|
Melanoma |
A malignancy originating from the melanocyte and now known to be linked to a variety of biochemical and genetic defects. Melanoma is an umbrella term for a variety of tumours with diverse biological behaviour. |
|
Merkel Cell |
Specialised cells in the epidermis of the skin that are important for proper neural encoding of light touch stimuli. |
|
Metastasis |
Spread of cancerous cells from their site of origin to other sites of the body. |
|
Mutation |
Heritable change in the nucleotide sequence of the DNA. |
|
Oncogene |
An altered gene whose product can acts in a dominant fashion to help make a cell cancerous. Typically, an oncogene is a mutant form of a normal gene (proto-oncogene) involves in the control of cell growth or division. |
|
Oxidation |
Loss of electrons from an atom. |
|
Reactive Oxygen Species (ROS) |
A reactive chemical species containing oxygen, such as the superoxide anion or hydrogen peroxide |
|
Tumour |
Abnormal mass of cells resulting from a defect in cell proliferation control. A tumour can be non-invasive (benign) or invasive (cancerous, malignant). |
References
Barckhausen, C., Roos, W. P., Naumann, S. C., & Kaina, B. (2014). Malignant melanoma cells acquire resistance to DNA interstrand cross-linking chemotherapeutics by p53-triggered upregulation of DDB2/XPC-mediated DNA repair. Oncogene, 33(15), 1964-1974.
Cancer Council NSW (2020). Skin Cancer Cancer Council NSW
Holíková, Z., Massi, D., Lotti, T., & Hercogová, J. (2004). Insight into the pathogenesis of sporadic basal cell carcinoma. International Journal of Dermatology, 43(12), 865–869.
Lucas, R. M., Yazar, S., Young, A. R., Norval, M., de Gruijl, F. R., Takizawa, Y., Rhodes, L. E., Sinclair, C. A., & Neale, R. E. (2019). Human health in relation to exposure to solar ultraviolet radiation under changing stratospheric ozone and climate. Photochemical & photobiological sciences : Official journal of the European Photochemistry Association and the European Society for Photobiology, 18(3), 641–680.
Nitzki, F., Becker, M., Frommhold, A., Schulz-Schaeffer, W., & Hahn, H. (2012). Patched knockout mouse models of Basal cell carcinoma. Journal of skin cancer, 2012, 907543.
Panich, U., Sittithumcharee, G., Rathviboon, N., & Jirawatnotai, S. (2016). Ultraviolet Radiation-Induced Skin Aging: The Role of DNA Damage and Oxidative Stress in Epidermal Stem Cell Damage Mediated Skin Aging. Stem cells international, 2016, 7370642.
Services, U.(2014). Skin Cancer As A Major Public Health Problem. [online] Ncbi.nlm.nih.gov.
The World Health Organization (2017). Ultraviolet (UV) Radiation And Skin Cancer.