DNA Repair Communiqué II
DNA – SOS
Introduction
The faithful transmission of our genetic code from one generation to the next places DNA – the source of our genetic material – at the centre of two fundamental problems we must overcome. First, cells must be able to perfectly replicate at each cell division. Second, cells must be able to identify and repair any damage once it has occurred. Exogenous factors – such as chemical exposure, ultraviolet light, and ionising radiation – and endogenous factors, such as reactive oxygen species, constantly threaten the stability of DNA and must be identified and restored immediately.
In DNA Repair Communiqué I, we outlined the fundamentals of cell division and DNA repair. The phases of the cell cycle outlined therein are fundamental to our understanding of the synthesis and replication of DNA. Furthermore, knowledge of how a cell repairs its DNA proteins, specifically the nucleotide excision repair (NER) pathway, is crucial to understanding key topics outlined below. Thus, we recommend reviewing DNA Repair Communiqué I before reading further if these concepts are not well understood.
Within DNA Repair Communiqué II we delve deeper into the topic of DNA synthesis and repair. We begin with the fundamentals of how DNA is synthesised, focusing on the role of DNA polymerases. Once this foundational knowledge is established, we review how cells cope with damage to the DNA template, using translesion DNA synthesis (TLS).
The Fundamentals of DNA Synthesis
As discussed in DNA Repair Communiqué I, the most basic function of the cell cycle is to duplicate human DNA into two genetically identical daughter cells through two overlapping, phases: nuclear division (mitosis) and the division of the cytoplasm to form two daughter cells (cytokinesis).
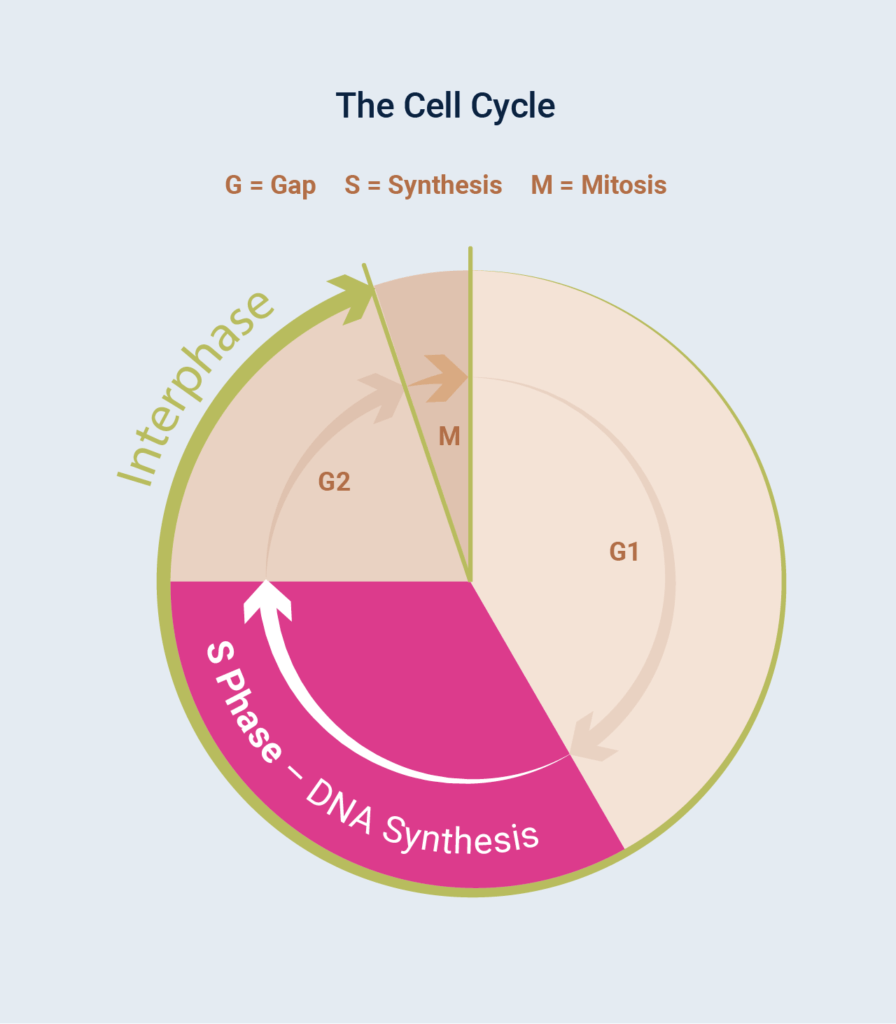
Although visually striking, the events of mitosis are only one phase of the cell cycle (Figure 1); for a typical mammalian cell, this period, known as M phase, usually lasts less than an hour. Cells spend most of their time in between divisions, a period known as Gap Phase, otherwise known as the G Phases.
Combined with S Phase and G2 Phase, this period is the longest growth period of the cell cycle, known as Interphase. During interphase, the cell is given time to grow and synthesise mRNA and protein required for the next step of the cell cycle. DNA replication occurs during S phase (S for synthesis), a part of the interphase, which requires 10–12 hours and occupies a significant part of the cell-cycle time in a typical mammalian cell.
![]()
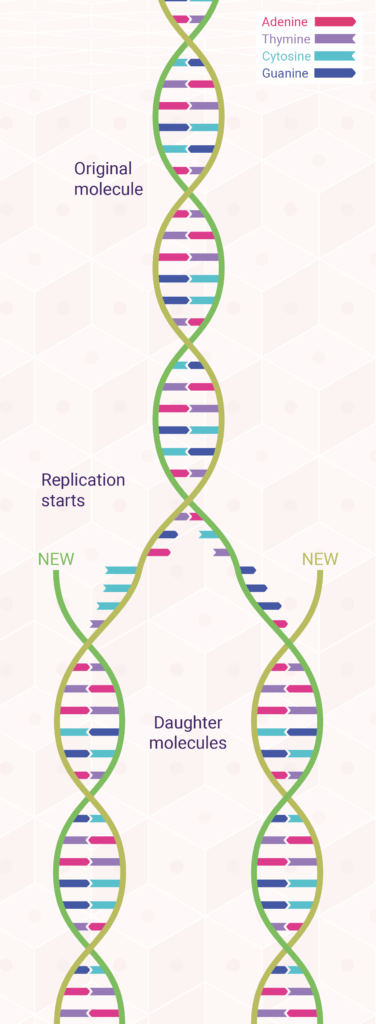
The underlying mechanisms of DNA replication depend on the double-helical structure of DNA. In fact, a month after Watson and Crick published their now-classic paper postulating a double helix for DNA, they followed up with an equally important paper suggesting how such a base-paired structure might duplicate itself.
The model Watson and Crick proposed for DNA replication was one of the two strands of every newly formed DNA molecule is derived from the parent molecule, whereas the other strand is newly synthesised.1,2 This is called semi-conservative replication (Figure 2) because half of the parent molecule is retained by each daughter molecule. Within five years of its publication, the Watson–Crick model of semi-conservative DNA replication was tested and proved correct by Matthew Meselson and Franklin Stahl, in collaboration with Jerome Vinograd.3
According to this model, during DNA replication, each strand of the DNA double helix serves as a template for the synthesis of a new complementary strand. As biologists proceeded to unravel the molecular details of this process, it gradually became clear that DNA replication is a complex event involving numerous enzymes and other proteins—and even the participation of RNA.
When the semi-conservative model of DNA replication was first proposed in the early 1950s, biologists thought that DNA replication would be so complex that it could only be carried out by intact cells. Just a few years later, however, Arthur Kornberg showed that an enzyme he had isolated from bacteria can copy DNA molecules in a test tube, work for which he received a Nobel Prize in 1959.4 This enzyme, which he named DNA polymerase, requires that a small amount of DNA be initially present to function as a template. More than a dozen different enzymes have subsequently been identified as being involved in the DNA replication and repair process.
Given the complexity of DNA replication, one might wonder how cells are able to accurately replicate DNA so often without causing errors. They cannot. About 1 out of every 100,000 nucleotides incorporated during DNA replication is incorrectly base paired with the template DNA strand, an error rate that would yield more than 120,000 errors every time a human cell replicates its DNA.5 One of the key roles of DNA polymerases is to remove incorrect nucleotides, improving the fidelity of DNA replication to an average of only a few errors for every billion base pairs replicated.
During replication, DNA synthesis stalls when replication machinery encounters a fault within the template strand. Persistent replication stalling and blockages can lead to cell death, as well as genomic instability, which may ultimately go on to form cancer if left unchecked. Thus, organisms have evolved DNA damage tolerance (DDT) mechanisms to bypass DNA lesions and enable cells to continue replication by incorporating the error into future strands of DNA.
Key to DNA replication, synthesis and repair is Proliferating Cell Nuclear Antigen (PCNA). PCNA works as a DNA clamp, encircling DNA during the replication process and acting as a scaffold in the recruitment of DNA polymerases. In this role, PCNA serves as a moving platform, recruiting various factors for replication or DDT, dependent upon which is required. Two of the most important polymerases PCNA interacts with are DNA polymerases delta (Pol δ) in DNA replication and DNA polymerase eta (Pol η) during TLS. A table of important DNA replication and repair proteins are included below for reference. More information on the specific functions of PCNA may be found in Scientific Communiqué VIII and Scientific Communiqué IX.
| Protein | Main Activities and/or Functions |
|---|---|
| DNA helicase | Unwinds double-stranded DNA |
| DNA ligase | Makes covalent bonds to join adjacent DNA strands, including the Okazaki fragments in lagging strand DNA synthesis and the new and old DNA segments in excision repair of DNA |
| DNA polymerase alpha (α) | Nuclear DNA synthesis: forms complex with primase and begins DNA synthesis at the 3˜ end of RNA primers for both leading and lagging strands (also functions in DNA repair) |
| DNA polymerase delta (δ) | Nuclear DNA synthesis; 3˜ S 5˜ exonuclease (for proofreading); involved in lagging and leading strand synthesis (also functions in DNA repair) |
| DNA polymerase epsilon (ε) | Nuclear DNA synthesis; 3˜ S 5˜ exonuclease (for proofreading); thought to be involved in leading and lagging strand synthesis (also functions in DNA repair) |
| DNA polymerase gamma (γ) | Mitochondrial DNA synthesis |
| DNA topoisomerase (type I and type II) | Makes single-strand cuts (type I) or double-strand cuts (type II) in DNA; induces and/or relaxes DNA supercoiling; can serve as swivel to prevent overwinding ahead of the DNA replication fork; can separate linked DNA circles at the end of DNA replication |
| Initiator proteins | Bind to origin of replication and initiate unwinding of DNA double helix |
| PCNA | Binds core polymerase subunit and keeps it on DNA |
| Primase | RNA synthesis: makes RNA oligonucleotides that are used as primers for DNA synthesis |
| RNA endonuclease (RNase H), RNA exonuclease (FEN1) | RNase H recognises RNA/DNA strands and nicks them; FEN1 then digests the RNA |
| Single-stranded DNA binding protein | Binds to single-stranded DNA; stabilises strands of unwound DNA in an extended configuration that facilitates access by other proteins |
| Telomerase | Using an integral RNA molecule as template, synthesises DNA for extension of telomeres (sequences at ends of chromosomal DNA) |
| XPV/ POLη | DNA-polymerase eta (pol-eta) performing trans-lesion DNA synthesis due to UV damage |
Translesion DNA Synthesis
The features that give DNA polymerases their high speed and accuracy (replication, recognition, and repair) also mean that they are easily stalled by DNA damage. If DNA lesions are unable to be fixed by nucleotide excision repair (NER), DNA synthesis is blocked as DNA polymerases cannot adapt to the damage effectively. These blockages can lead to the collapse of the replication fork, compromising DNA synthesis and potentially causing severe consequences such as the formation of cancer. Considering this risk, nature has equipped the human body with several alternative pathways to tolerate (rather than fix) DNA damage using specialised DNA polymerases in a process known as translesion DNA synthesis (TLS). Within this section, we shall focus on this complex process through the lens of XPV protein, also known as DNA polymerase eta (Pol η). In humans, the XPV protein, encoded by the XPV gene, is responsible for bypassing a very common form of sunlight-induced damage, cyclobutene pyrimidine dimers (TT dimer(s)), and is deficient in patients with XP-variant. Subsequently, these patients are at a far higher risk of non-melanoma skin cancers as the body is unable to tolerate the damage as effectively, leading to severe consequences to each damaged cell.
TLS is a DNA damage tolerance process that allows the DNA replication machinery to bypass damage to the DNA template strand. Whereas most translesion synthesis polymerases have a low fidelity (error prone), XPV protein is believed to have a far greater level of accuracy.
TLS is one of the most complex processes within DNA repair and is yet to be fully understood. The process may, however, be simplified as a trade-off. When a blockage occurs within the DNA strand during replication, the cell has a choice: it can stop the replication process, thus risking chromosomal stability within the cell and likely resulting in cell death. From a cellular perspective, enabling the pyrimidine dimer to remain is preferable to allowing the TT dimer to remain or attempt to repair the dimer in situ, both of which could lead to severe consequences for our genetic stability.
Because the TT lesion has not been eliminated, TLS is a damage tolerance mechanism, enabling damage to remain. This process has subsequently become known as an SOS pathway as it is only initiated once all alternative mechanisms of repair have failed.
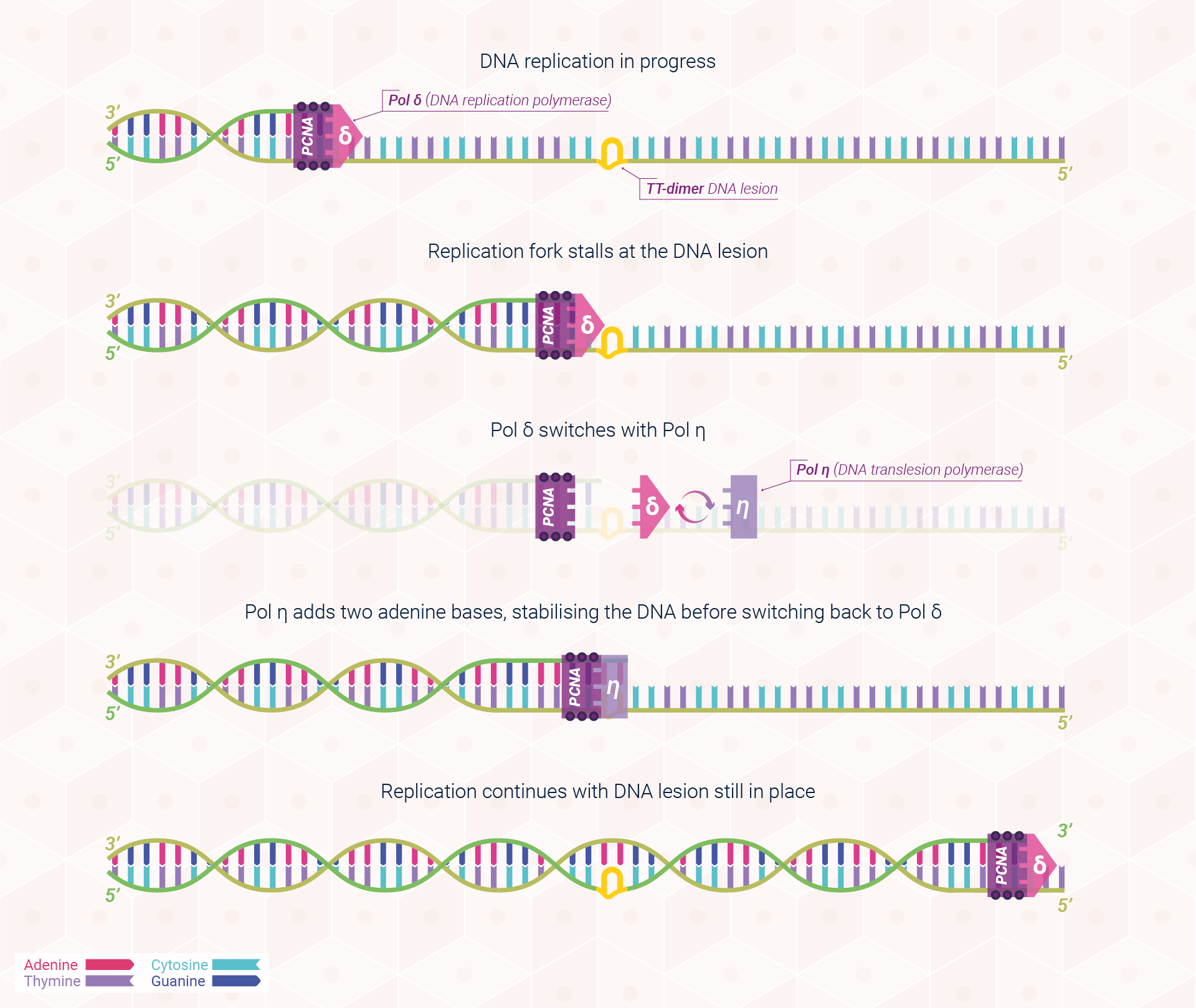

Thus, following the presence of a TT dimer in the replication fork, TLS enables DNA replication to continue through switching normal DNA polymerase for DNA polymerase eta, which instructs the DNA strand on how to bypass the lesion and allow cell proliferation, and thus human life, to continue. Figure 3 provides a simplified visualisation of this process.
XPV and Translesional Synthesis
Xeroderma pigmentosum (XP) is a rare autosomal recessive human disorder that is characterised by extreme sensitivity to ultraviolet radiation. While most XP patients exhibit a defect in nucleotide excision repair, about 20% of patients, the so-called XP-variant (XPV), have a normal excision repair system but are defective in their capacity to replicate lesion containing DNA using POLη.
Although often referred to as a milder subtype, XPV patients still have a far greater risk of non-melanoma skin cancers compared to the general population. The lack of XPV protein leads to a strong blockage of the replication forks in XPV cells, thus causing severe abnormalities within the cell leading to an increased risk of cancer.
While replication can still recover through the action of other alternative DNA repair pathways and polymerases, these polymerases are less efficient in their repair and thus carry the risk of causing point mutations within cells and increasing the risk of oncogenesis. Regrettably, XPV patients are thus forced into a life of sun and ultraviolet light avoidance and systemic photoprotection as a therapy is still lacking.
Concluding remarks
All living organisms on the planet are subjected to the continual threat of DNA damage. Resultantly, nature has equipped us with a plethora of DNA repair pathways to protect ourselves from the long-term consequences of such attacks. Unfortunately, situations nevertheless arise where damage is not correctly repaired, and cells are forced to adapt and overcome.
Translesion DNA synthesis (TLS) provides an SOS pathway for tolerating irreparable DNA damage. Through specialised DNA polymerases, our body may continue cell proliferation and thus maintain homeostasis, though at a cost. Damage to template strands cannot be repaired and thus the introduction of point mutations within our body increases the collective risk of cancer, especially if the number of mutations is large, or situated next to an oncogene (such as P53).
Leveraging alternative pathways of DNA repair and assisting the body in DNA damage recognition can mitigate the required risk of using SOS pathways. Within the skin – the first line of defence against environmental threats such as ultraviolet radiation – the MC1R signalling axis has shown promise in helping to reduce the risk of oncogenesis and ultimately skin cancer; a topic we will explore further in DNA Repair Communiqué III.
References
- Watson, J. D., & Crick, F. H. (1953). Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid. Nature, 171(4356), 737-738.
- Watson, J. D., & Crick, F. H. (1993). Genetical implications of the structure of deoxyribonucleic acid. JAMA, 269(15), 1967-1969.
- Meselson, M., Stahl, F. W., & Vinograd, J. (1957). Equilibrium sedimentation of macromolecules in density gradients. Proceedings of the National Academy of Sciences of the United States of America, 43(7), 581.
- Kornberg, A. (1960). Biologic synthesis of deoxyribonucleic acid. Science, 131(3412), 1503-1508.
- Hardin, J., Bertoni, G. P., & Kleinsmith, L. J. (2017). Becker’s World of the Cell, eBook. Pearson Higher Ed.
- Swope, V., Alexander, C., Starner, R., Schwemberger, S., Babcock, G., & Abdel‐Malek, Z. A. (2014). Significance of the melanocortin 1 receptor in the DNA damage response of human melanocytes to ultraviolet radiation. Pigment cell & melanoma research, 27(4), 601-610.
- Abdel-Malek, Z. A., Swope, V. B., Starner, R. J., Koikov, L., Cassidy, P., & Leachman, S. (2014). Melanocortins and the melanocortin 1 receptor, moving translationally towards melanoma prevention. Archives of biochemistry and biophysics, 563, 4-12.
- FitzGerald, L. M., Fryer, J. L., Dwyer, T., & Humphrey, S. M. (2006). Effect of MELANOTAN®,[Nle4, D-Phe7]-α-MSH, on melanin synthesis in humans with MC1R variant alleles. Peptides, 27(2), 388-394.
- Minder, E. I., Barman-Aksoezen, J., & Schneider-Yin, X. (2017). Pharmacokinetics and pharmacodynamics of afamelanotide and its clinical use in treating dermatologic disorders. Clinical pharmacokinetics, 56(8), 815-823.
DNA Repair Communiqué Library
The DNA Repair Communiqués provide an in-depth look
at the work we conduct and the rationale for developing melanocortins.
Glossary
- AP endonuclease
- A multifunctional enzyme involved in base excision repair (BER).
- Base Excision Repair (BER)
- A repair mechanism that corrects DNA damage from oxidation, deamination, and alkylation.
- Cell cycle
- The series of events that take place in a cell that cause it to divide into two daughter cells.
- Chromatin
- A specialised form of DNA contained within the cell nucleus.
- Chromosome
- The part of the cell that houses the genetic material.
- Cytokinesis
- The process by which one cell physically divides into two cells.
- Deaminated base
- DNA in which an amine base has been lost (cytosine and thymine).
- Depurinated base
- DNA in which a purine base has been lost (adenine and guanine).
- DNA glycosylase
- A family of enzymes involved in base excision repair,
- DNA polymerase
- An enzyme that synthesises DNA by joining nucleotides together using a DNA template as a guide.
- Global genome repair (GGR)
- A subtype of NER which repairs UVB induced lesions in the non-transcribed regions of the DNA.
- Interphase
- Interphase is composed of G1 phase (cell growth), followed by S phase, followed by G2 phase (cell growth), after which the mitotic phase begins.
- M phase
- The time-period in which the cell divides into two cells (parent and daughter cell).
- Mitosis
- A process of cell duplication, or reproduction, during which one cell gives rise to two genetically identical daughter cells.
- Mitotic spindle
- A highly dynamic molecular machine, which assembles around the chromosomes and distributes the duplicated genome to the daughter cells during mitosis.
- Nucleotide Excision Repair (NER)
- A process that repairs damage to one strand of the DNA, particularly from UV irradiation, which distorts the DNA helix.
- Oncogene
- A gene that is a mutated (changed) form of a gene involved in normal cell growth. Oncogenes may cause the growth of cancer cells.
- Proliferating cell nuclear antigen (PCNA)
- PCNA is a DNA clamp that acts as a processivity factor (assisting DNA in carrying out continuous synthesis) by acting as a scaffold to recruit DNA polymerases onto the replication fork and strands.
- Pyrimidine Dimer
- A DNA mutation formed because of UVB radiation exposure.
- S phase
- The time-period in which DNA replication occurs and the DNA molecule is duplicated.
- Semiconservative replication
- A hypothesis for cell replication wherein the DNA double helix unwinds, with each parent strand serving as a template for the synthesis of a complementary daughter strand.
- Sister chromatids
- Two identical chromatids that are formed by replication of a chromosome during the S phase of the cell cycle.
- Transcription-coupled repair (TCR)
- A sub pathway of NER, it removes lesions from the template DNA strands of actively transcribed genes.
- Translesion DNA synthesis (TLS)
- An SOS pathway initiated once all other methods of repair have failed and damage is permanently located in the template strand. TLS enables the DNA to bypass damage in these instances by copying DNA containing unrepaired damage into the new DNA strand during replication.
- Xeroderma pigmentosum (XP)
- XP is a rare autosomal recessive disorder of DNA repair characterised by one of eight defective enzymes within the XP gene, involved in DNA repair