Variegate Porphyria
About VP
The porphyrias are a group of nine genetic metabolic diseases, including VP and EPP, characterised by enzyme deficiencies along the biochemical pathway of haem synthesis. VP is a rare autosomal dominant inherited disorder resulting from the deficiencies of the mitochondrial enzyme photoporphyrinogen oxidase (PPOX), the sixth enzyme in the haem biosynthesis pathway.
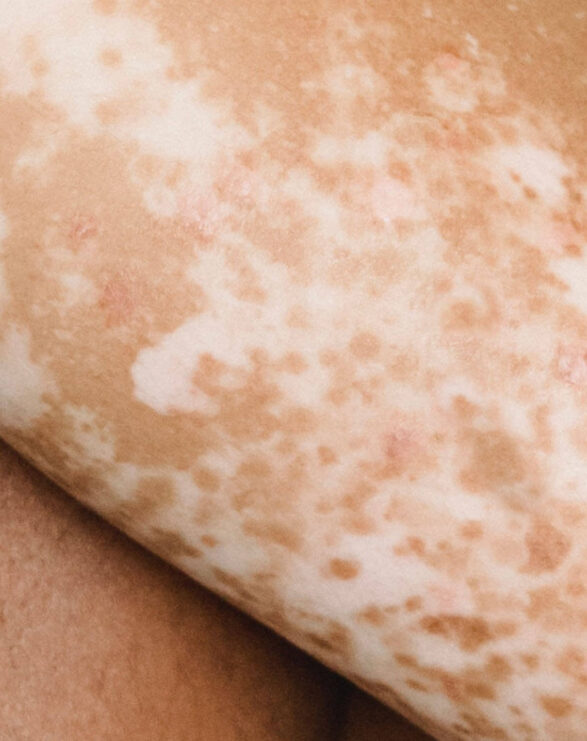
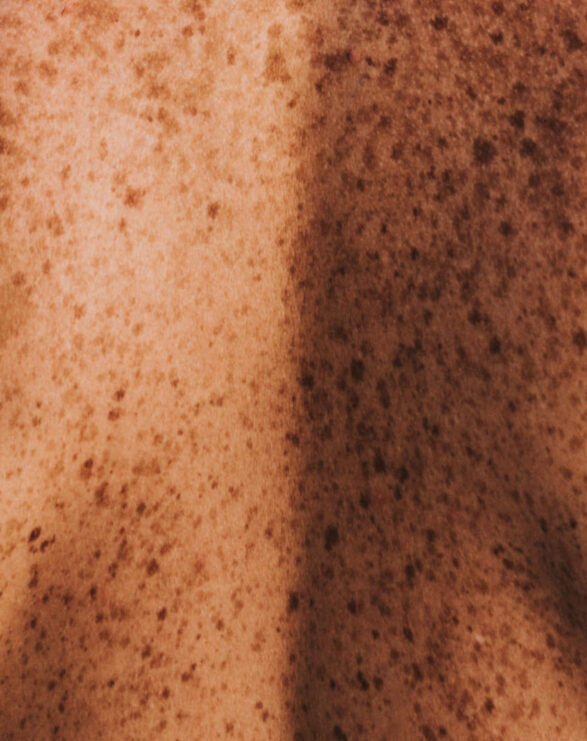
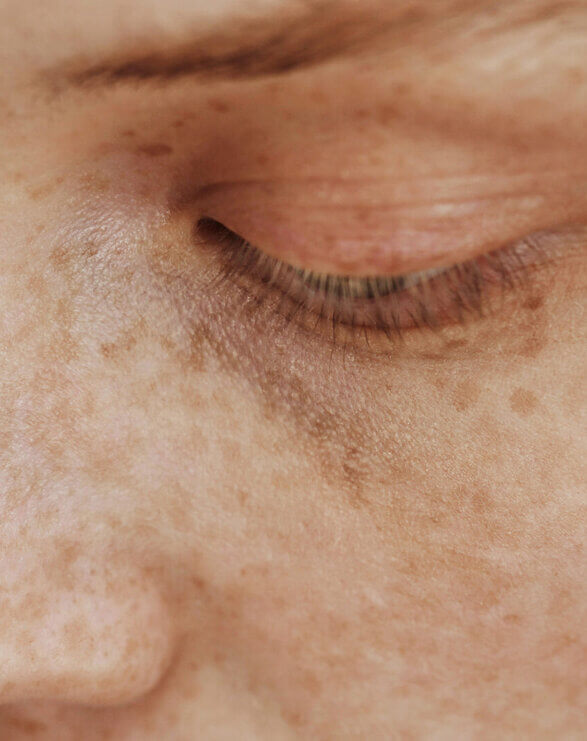
A debilitating and burdensome disease for patients, VP is classified as both a cutaneous and acute porphyria. The cutaneous symptoms are characterised by adult-onset blistering lesions (bullous symptoms, subepidermal vesicles) and chronic fragility (hypotrophy and hypoplasia) of sun and light-exposed skin, especially the back of the hands and the face. The propensity of the dermis to form wounds (lesions and erosions which form crusts, heal slowly or not at all, and are prone to infection) following the mechanical use of hands and feet further restricts VP patients in their daily activities.
Typically, patients experience scarring of the face, hands and feet. Patients diagnosed with VP experience phototoxicity and suffer episodes in spring and summer when the atmospheric intensity of light increases. The acute neurovisceral symptoms may present as severe episodes as early as adolescence, with women more affected than men. Psychiatric disturbances, autonomic neuropathy and muscle weakness have been reported in some patients.
The reported prevalence of VP varies from 0.3 to two cases per 100,000 inhabitants but has been reported as more common in the Caucasian South African population (up to one in 300 people) due to a founder effect. European prevalence varies from 0.32 to one case per 100,000 inhabitants, whereas the reported prevalence in the US is 0.5 case per 100,000 inhabitants.
Currently, there is no standard of care or alternative therapy for cutaneous VP symptoms. Patients are forced to lead a life sheltered from light and to avoid any mechanical contact with their skin surface or risk triggering wounds and long-lasting blistering.
Clinical & regulatory progress
June 2024
CLINUVEL’s drug afamelanotide received a positive opinion for an orphan drug designation (ODD) from the European Medicines Agency (EMA) for the treatment of variegate porphyria (VP). Read the announcement.
March 2024
CLINUVEL announced positive results from its first Phase II study evaluating SCENESSE® (afamelanotide 16mg) as a treatment for variegate porphyria (VP) patients. Read the announcement.
May 2023
CLINUVEL announced the commencement of the new clinical study (CUV040) evaluating the safety and efficacy of SCENESSE® in VP patients. Read the announcement.
October 2018
CLINUVEL announced that it has reached agreement with two European porphyria expert centers on a clinical trial protocol to conduct a Phase IIa proof of concept study evaluating the safety and efficacy of SCENESSSE® (afamelanotide 16mg) in variegate porphyria (VP).
References
Elder G, Harper P, Badminton M, Sandberg S, Deybach JC. The incidence of inherited porphyrias in Europe. J Inherit Metab Dis. 2013;36(5):849-857. doi:10.1007/s10545-012-9544-4.
Horner ME, Alikhan A, Tintle S, Tortorelli S, Davis DM, Hand JL. Cutaneous porphyrias part I: epidemiology, pathogenesis, presentation, diagnosis, and histopathology. Int J Dermatol. 2013;52(12):1464-1480. doi:10.1111/ijd.12305.
Singal AK, Anderson KE. Variegate Porphyria. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; February 14, 2013