CLINUVEL Newsletter
Dear shareholders, friends,
As a new world presents itself indicating that the virus will not easily be eliminated as we are accepting to find a modus co-existentiae, albeit we need to pause for the more than 600,000 people who have lost their lives and countless more impacted by this pandemic.
For CLINUVEL, as iterated in the May News Communiqué III, we have looked at changing operations as an adaptive response. In this Communiqué IV, we think about the emerging world and some of the key learnings related to our business. A paradox emerges: we find ourselves in an era of restricted activities, widespread anxiety about future outlook, and general slowdown in pharmacological activities, but at CLINUVEL we are, – perhaps more than at previous times – confident in the value we see coming from our R&D projects and commercial activities in Europe and US.
Over the past quarter, central banks and governments have stepped up fiscal intervention to avoid a lack of liquidity in the markets. Like we saw in 2007, interest rates have fallen further with a central effort to reignite economies by disincentivising savings on deposits. As the first stimulus appeared to wane, quantitative easing became the next instrument used by the FED, ECB and Bank of Japan as principal buyers turned to long-term treasuries and corporate bonds. Equally, other paper and the provision of bank loans are making up the total stimulus packages globally. From a wider perspective, in News Communiqué III we referred to the parachute rescues to households and those losing their jobs. In Australia, the Jobseeker and Jobkeeper schemes are aiming at both employers and employees when financial support is needed. In addition, the Australian Tax Office has put in place a Cash Flow Boost for companies eligible to receive a tax-free payment. Singapore has a Jobs Support Scheme in place contributing towards the gross monthly wages paid to local employees.
Within the European Union, a deep divide is seen between political leaders as the richer, frugal countries refuse to assist the shortfall of the southern Mediterranean countries. As this document goes to publication, heated debates are being reignited in Europe as some of the leading nations argue for a €750 billion economic recovery package to help European countries offset the damage wrought by COVID-19. The Netherlands contends vehemently that such a plan would provide countries with unprecedented grants lacking the necessary surveillance. In the US, the debate between Democrats and Republicans about further stimulus has not reached a similar climax since the House of Representatives has already conceded to more than US$2 trillion in stimulus.
In the UK, Chancellor Sunak’s award of £1,000 “furlough bonus” for businesses which retain their employees beyond January 2021 is only one of the many measures to aid companies and employees during the summer. The total spending related to COVID by the Johnson government has been north of £350 billion and with an additional £100 billion expected for the current financial year. No doubt this will eventually lead to an increase in taxes, be it income or corporate taxes, in spite of the manifesto issued by the Tories at the time of the last elections. However, VAT rates in the UK have been temporarily reduced from 20% to 5% on tourism and hospitality-related activities.
Despite the collective support, there have been massive layoffs across the world. US unemployment was cited by the Bureau of Labor Statistics to have improved over June totalling 11.1% or 17.8 million unemployed. In total 141.2 million people are currently employed in the US, be it full or part-time, translating to a ratio of 12.6% in real unemployment.
On 07 August, we will receive the July figures from the US to assess the ongoing effects of COVID on unemployment. In comparison, in Germany we see 44.6 million people employed and 1.93 million unemployed: a total unemployment rate of 4.4%. The low numbers in Germany can be attributed to an efficient establishment of the “Kurzarbeit Programme” establishing short-term employment protection facilities. At the end of Q1 this year the UK recorded 3.9% unemployment, but then again, the Bank of England has caused wide-spread uncertainty by predicting a 10% unemployment rate by end of 2020. In the Euro zone an overall 7.3% unemployment has been estimated, while claimants on social benefits are rising sharply. In Australia, the adjusted unemployment rate increased to 7.4% in June. All these numbers should cause us to stand still and reflect as they are a catalyst for structural changes to our societies for years to follow.
My longer-term concern centres on our collective efforts to keep people away from both each other and work, as it will compound the economic slowdown. In this scenario, I fear that we will enter a faster conversion from a deflationary environment to a rapidly rising inflation rate post-COVID. As most economists would have agreed earlier this year that inflation was not a conceivable idea, there are more voices daring to contemplate a surge in prices.
Although many central banks within the Eurozone strive to aim for a target of 2% inflation, I am no longer sure this target is realistic. The realities of daily transactions are that prices are on the rise in specific sectors, contrary to the wide-held belief that inflation is absent. We are compelled to think about the implications for CLINUVEL and all the measures we can take to prepare to manage various scenarios ahead of time.
As stated, from past years, I view fiscal deficits, spending spikes and public debt as the main risks to the stability of our ‘modern’ economy. The current climate is once again one where widespread lending by banks is promoted to keep businesses afloat, on one hand applaudable and on the other hand unsustainable, since lack of cashflows impacts the ability to service these debts let alone paying off the principal; I foresee that many companies will be at peril. From our perspective, we have consciously kept CLINUVEL debt-free and away from additional financial risks, despite the low interest rates on offer, particularly in times when economic uncertainty rules.
Geopolitical issues have been my focus of attention, noting the deterioration of Sino-American trade and the decline of the Sino-Australian (trade) relations. It was only a few years ago, in March 2015, that China and Australia signed a free trade agreement. The geopolitical dynamics are of high relevance to us as we try to calculate the short and mid-range permutations and to discern new opportunities.
For the softer and not unimportant aspects of this all, I see the loss of morale and widespread disillusion among young aspiring professionals and changing demand for skills as a significant challenge to both the labour market and real issues in our economies. Our interconnectedness means that effects are not only felt across continents and sectors, but also across the various subsidiaries we operate. That said, within the Group, I see it as our task to increase productivity and preserve employment throughout adverse conditions, and ask from our staff that they commit to a stricter discipline while working from home to keep their eye on meeting both annual and longer-term (three year) corporate objectives. At the same time, I regard it as our duty to accompany the next generation of management leaders and guide young professionals through unprecedented times by offering work security. In return, I do wish to see superior performance and maximum use of skills and time. Eventually, this showing of corporate loyalty will be reciprocal (if it is not already).
In terms of implant supply, prescriptive treatment provided, productivity, and technological progress, the teams have done better than we could have hoped for and we are reasonably content with the overall output as the second lockdown in Australia and other parts of the world is underway.
Our management and Board have frequently published an aligned view on business metrics, such as cost control, cash positivity and overall profitability to navigate through turbulence and economic uncertainty in markets. Although short term gain is important to us, I see it as my first duty to focus on a longer horizon and planning how CLINUVEL will emerge from the economic crisis.
At operational level, we are united by a set list of short and midterm performance objectives which have been highlighted at the November 2019 Annual General Meeting, and – inch by inch – we are moving towards these goals as best seen from the progress the past two calendar years (refer to the Public and Investor Relations section below). The success we have achieved in our sector has come in the form of a ‘dancing procession’, two steps forward and one back, and an inventory of the calibre of professionals within the Company gives me confidence that the diverse mix of talent will deliver on new challenging objectives we set ourselves.
There is a clear direction towards where CLINUVEL will be in two years’ time and, so to speak, we are working on a construct wrapped in a temporary scaffold. As we are building the permanent layers, new technical data are coming through, CLINUVEL’s portfolio of melanocortin products is emerging and our aim is on record to be a diversified house to lead on melanocortins and specialty drugs. Clinical value is being established while enterprise value will follow.
In the case of melanocortins coming to the fore globally, it appears that the time is right for this technology to mature. From our early analyses, “the directional gamble” CLINUVEL took in 2005 has had considerable merit and this provides the confidence to continue developing proopiomelanocortin (POMC) molecules for future use. In November, I communicated that it is rare that ‘novel’ technology survives three decades, most frequently technology becomes obsolete within 15 years for superior science to emerge. In the case of melanocortins, the reverse is true: as time passes there is more relevance found from adjacent fields of science to justify the use of POMC technology in a broader range.
USA DISTRIBUTION UPDATE
The number of US centres trained and competent to administer the SCENESSE® (afamelanotide 16mg)1 treatment is increasing at a satisfactory rate, while all of the prescribing physicians have proven apt at administering the drug subcutaneously (underneath the skin). Generally speaking, for dermatologists the implant administration underneath the skin above the hipbone (supra-iliac crest) is a new procedure requiring some dexterity. For those who are used to taking frequent biopsies and performing Mohs surgery (a type of skin cancer surgery), the SCENESSE® procedure proves simple and without concerns. For others with less experience, CLINUVEL’s training and videos prove to be great help.
Our teams often receive queries as to whether the implant administration requires a scalpel, incision, and sutures. The answer is negative, SCENESSE® is administered through a trocar injection under continuous manual pressure and no incision or closure of the puncture site is required. Patients receive a small band aid or dressing and typically leave the clinic after 30 minutes of observation. In the US, the frequency of dosing is six implants per annum, and it appears that the majority of US patients will be requesting treatment all year around.
As part of the start of US distribution, our cross-functional team is systematically engaging with the US insurance providers allowing Prior Authorization (PA) for US erythropoietic protoporphyria (EPP) patients. We refer to the mechanism of PA in the announcement made on 23 March.
The US pricing of SCENESSE® under “medical benefit” is uniformly being adopted while the system of institutional processing of the reimbursement is variable across different states whereby generally payment terms range from 60 to 120 days. Each state bound insurer typically has a contract or arrangement with the local Specialty Center (“within network”), and after agreeing PA and treatment codes, the insurer reimburses the treatment center for the SCENESSE® implant and administration procedure. After the Specialty Center has received the insurance funds, it in turn reimburses CLINUVEL for the cost of the drug. This cycle of receiving funds is proving to take longer than is the case in the European Economic Zone.
Thus far, only one physician has been requested to resubmit for PA, while all other prescribers have received approval to administer SCENESSE® within two weeks after submission.
The number of insurers agreeing to PA has strongly increased keeping pace with the number of patients signed up to our program seeking treatment reimbursement since April and the number of Specialty Centers spread across the US is surpassing our projections. The speed of ordering and drug administration relies on the turnaround of approvals by each insurer per state. In some of the larger Centers, the final paperwork is awaited for the inclusion of SCENESSE® in the local institutional formulary. All in all, we are executing ahead of our planning.
EXPANSION OF CLINUVEL
In these times when supply chains and hospitals are slowing down or freezing the conduct of clinical trials, naturally we have been discussing how to maintain the canter and progress of CLINUVEL. In these times of adversity, compact teams like CLINUVEL can demonstrate resourcefulness and creativity in solving unexpected problems. Our growth is set against the range of effects of the pandemic providing for different scenarios.
We view the expansion of the Group as essential and, as stated in past years, this will stem from both organic forms of projects, products and clinical applications, while the inorganic part will aim to vertically integrate services and functions. The past years we have seen the costs of manufacturing, labelling, printing, packaging, medical and OTC product branding gradually increase as dictated by third parties. This comes with the large chains in pharmaceuticals, and our focus is to bring most functions under one roof to control speed of process as well as costs.
I expect more of these activities to materialise in the coming months, despite the global slowdown. I view diversification as welcome, or in street wise terminology, whatever it takes to build a solid business on several pillars. Although, CLINUVEL is at present in an optimum economic state, I do not regard it as a comfortable position and more activities are needed to grow the Group.
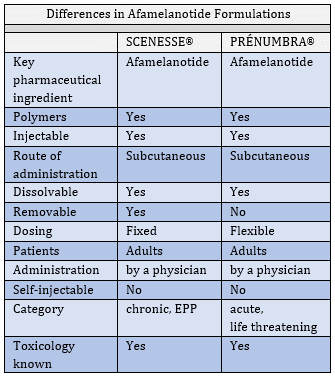
SCENESSE® VERSUS PRÉNUMBRA®
In July, we revealed our second systemic pharmaceutical product in development. With PRÉNUMBRA® we are progressing a new formulation as part of the life-cycle management of afamelanotide. There is a clear sequence to follow, and now that the regulatory agencies accede to the safety profile of afamelanotide and accept the introduction of a novel hormone analogue, we broaden the technical dossier to additional pharmacological modes of action and applications of the molecule. In this technical endeavour it is essential to take the regulatory decision makers along. In regular dialogues and formal additions to the New Drug Application (NDA), one seeks expansion of the pharmacology modules (formally written sections) to justify how a new molecule would affect different organs and receptors. In lay terms, translation of a medicinal product requires continuous discussion with regulatory authorities to minimise surprises and resistance.
The sequence of timing was essential, first European followed by US approval, Australian progress and – gradually – the expansion in systemic diseases which remain yet untreated. Following years of more than 20,000 data points of analyses, the time had come for progress into a non-solid dose formulation, PRÉNUMBRA®. The ultimate evidence to use new formulations comes from in vitro data generated in our laboratories, pre-clinical studies (limited to the regulatory minimum required) and human exposure. Sometimes, one discovers new data from dosing strengths, frequency of administration, and (inevitably) from side effects of a drug, and together these data provide progressive insights summating years of work.
At CLINUVEL, we sought to expose as many different disease populations as possible to gather data on the pharmacological profile and side effects of SCENESSE®. These data are invaluable and part of CLINUVEL’s specific knowhow providing a competitive position over others who are trying to emulate our programs. Our scientific teams have gradually acquired knowledge on the optimum therapeutic windows for a liquid controlled-release injectable formulation to be used in a number of diseases which either lack an effective therapy or where existing therapies provide too many side effects.
The main clinical objective is to make afamelanotide available in three further disorders where considerable clinical gain is to be expected for patients and where, eventually, a commercial opportunity exists. For these programs we aimed to use a liquid pharmacological product while shortening the length of clinical trials, whereby two key considerations dominate:
- the toxicology and safety from the use of afamelanotide had been well proven after two decades of use; and
- existing clinical data from the SCENESSE® implant count towards the building of a regulatory dossier (marketing dossier) for PRÉNUMBRA®.
In these upcoming trials sponsored by the Company – as opposed to physicians’ initiated trials – we start with a set of realistic endpoints (clinical objectives) to demonstrate minimally important difference; that is a clinical benefit to patients which genuinely helps them in their prevention or management of the disorder. The value of a new medicinal therapy lies in its clinical relevance and acceptance by payors and insurers.
In PRÉNUMBRA®, we aim to establish a formulation which allows higher and flexible dosing in acute and life- threatening conditions. Whereas, in PRÉNUMBRA® there is now an ability to use a liquid dose which is administered underneath the skin (subcutaneously), with the SCENESSE® implant there is no opportunity to adjust the dose when it is needed. The non-solid formulation will release the active drug afamelanotide in a specified manner tailored to treat particular diseases.
We keep emphasising the maintenance of a flawless safety profile in the evolution of CLINUVEL. In other words, without the highest degree of confidence that the active substance would remain safe and tolerable over a long period of time, we could not proceed to a higher dose in treating acute disorders. Without this safety profile there is, in reality, no further investment warranted. Our latest detailed analyses in Q1 2020 was based on 15 years of continuous data on the use of afamelanotide which provides the final threshold for making the liquid formulation available. Although, one can never be totally certain about adverse reactions to a drug, as part of technological risk management we have arrived at a point where causality between drug and mild side effects is consistent and sufficiently known for regulatory authorities to acknowledge the 20 years of exposure, translated in technical terms to more than 30,000 days of patient exposure coming from more than 10,000 dose administrations.
The patent life of new pharmaceutical products requires active management, therefore one wishes to see the start of the patent clock running as late as possible to ensure maximum patent life. Intellectual property in pharmaceuticals has been the subject of an intense debate in western parliaments since there are two camps arguing the positions of pharmaceutical enterprises monopolizing treatment options. Our views have not changed over the years on protection of our intellectual property. At the end of the day pharmaceutical companies which are actively investing in technologies for untreated diseases deserve longer term protection as incumbents. This is even more pressing when the funding is entirely from financial markets with the aim of finding innovative solutions. Governments can assist if and when they share in the risk upfront and early on, but scrutinising expenditures post-factum is just a comfortable but unrealistic debate across reclining chairs.
In the US, we are seeking further patent extension for SCENESSE® under the Hatch-Waxman Act to compensate for the length of the FDA approval process.
PUBLIC AND INVESTOR RELATIONS
The ongoing coronavirus pandemic also reinforces the importance of company announcements to keep stakeholders around the world informed on a timely basis on the progress of the Group.
We mentioned in News Communiqué III that CLINUVEL personnel in investor relations and public affairs were adopting available technology to communicate effectively with stakeholders, as face to face physical meetings have become too risky to personal health due to the coronavirus pandemic.
The increased activity of the Company over the last two years in the performance of its strategic initiatives is seen from the rise in the number of company announcements to the Australian Securities Exchange (ASX). From 44 announcements in the year ending June 2018, the number rose to 62 and 67 in the years ending 30 June 2019 and 2020, respectively. Since January 2020, the key announcements and updates have been:

These announcements are available on the CLINUVEL website, www.clinuvel.com. The frequency of News Communiqués and Chair’s Letters (six in total, so far this year), is a distinguishing feature of CLINUVEL’s communications approach, with the Managing Director and Chairman committed to regular and direct communication to stakeholders throughout the year.
CLINUVEL’s shareholders are dispersed around the world. The most recent data of June on beneficial shareholdings indicate the largest geographic shareholder concentrations are in Europe, North America, and Australia, in that order. The composition of shareholders by type of shareholder is also changing due to increasing institutional interest which has driven a recent rise in North American based shareholders. The challenge to CLINUVEL is to ensure updates on its progress are provided to shareholders on a timely basis, both directly and through multiple communication channels across the globe, in line with the requirements of relevant regulators and the needs of the Group. Upon an announcement to the ASX our communications team ensures the news is distributed via the CLINUVEL email updates list, is posted to the Group website (www.clinuvel.com) and social media channels, and is disseminated to worldwide media outlets. In general, and perhaps less commonly seen, the Company also puts out media announcements in simpler terms for a wider audience to understand the less technical publication.
As with recent communiqués, this Communiqué IV will be translated in German and posted on the website as promptly as possible for the convenience of German, Swiss and Austrian shareholders. The feedback we receive on this as a further initiative to communicate more extensively is appreciated
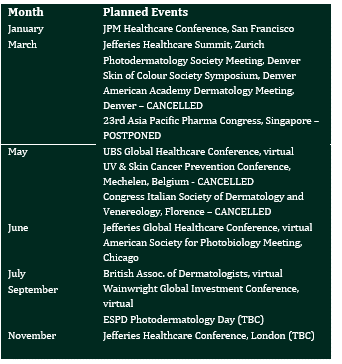
The calendar of planned events is updated, noting the prevalence of our participation in virtual conferences since the onset of the coronavirus pandemic, when these events have not been cancelled.
In the coming weeks, we will be announcing the aforementioned June quarter 2020 Cash Receipts and the annual financial results for the year ending 30 June 2020 will be released by the end of August. We will subsequently complete the 2020 Annual Report and prepare for the Annual General Meeting to be held in November.
SHORT SELLING
We have received questions on the continuous short positions against CUV (ASX) and CLVLY (Level I American Depositary Receipts traded over-the-counter and sponsored by the Bank of New York Mellon). As we speak, the shorts registered against CUV (ASX) make up 8.00% of total issued shares, after peaking at 9.65% in April 2020.
Management does not see it as its task to provide an explanation of share trading and short selling, however, a number of characteristics of this activity in respect of CLINUVEL have stood out. For the sake of providing factual information to supportive shareholders who have invested both in the Company and the management team, some words on this topic are warranted. We recognise how frustrating it is for some of the long-term shareholders to see the share price fluctuate and drop, as has occurred since October 2019. On the other hand, we note some shareholders see it as a welcome opportunity to invest in CLINUVEL based on its fundamental performance.
In the stock market, the practice of “shorting and distorting” is a well-known activity amongst brokers and market speculants, knowing that gains can be made by spreading false and negative news about a company. By persisting in this behaviour, the hope of market disruptors is that current shareholders will eventually become anxious and sell their stock, driving the price down even further. In a period whereby profit taking is expected, for instance when CLINUVEL achieved its epic success by obtaining FDA approval on 08 October 2019, it was quite easy to gamble against a company’s stock price by finding parties willing to lend out stock.
Historically, the rise in short selling of CLINUVEL shares started in April 2019, and in retrospect the newsflow on CLINUVEL has been very positive; there is absolutely no reason to currently hold that the Company will cease to perform. Management and the Board are motivated, progress is being made both commercially and in further development of new products and indications, and the financial health of the Group is excellent. There is a wide held belief in our field that this is the most driven and ambitious team one could possibly find among many Asian-Pacific life science companies.
Of course, the shock movement of economic plates exerts its influence on all businesses including CLINUVEL. While the stock markets have performed surprisingly well and many companies are gaining access to capital as it is needed, at CLINUVEL we have seen some unusual trading patterns the past months.
The Company will continue in the same mode and is fully aware of the concerns of its benign shareholders who are investing and supportive of the Company and its direction. However, the Company distances itself from those who disseminate negative and disparaging news on us. In this environment, some market participants see the opportunity fit to distort and short stocks within the gaze of regulatory oversight. The Australian Securities and Investment Commission has oversight of market trading in Australia and we at CLINUVEL play a role to point Australian, German, and US authorities in the jurisdictions where our shares are traded in the direction of these market participants and stock bashers and all those who distort the CLINUVEL narrative openly. Our duty is to protect shareholder value, and in these uncertain times we see it even more pertinent for financial regulators to step up and trace these individuals where it is needed.
Often, the short sellers infiltrate communities of retail shareholders online and influence news flow among brokers by distributing incorrect and often false rumours on the targeted company: the distorted news spread by short sellers mostly follow common themes varying from critique on strategic direction, spending and costs, overall share value, lack of expected returns, calibre of management and preference for other exchanges. During these periods, short sellers aim to discourage existing and new shareholders to remain in or enter a stock. The practices are widespread among many exchanges and eventually financial regulators step in, track and hold the culprits accountable and take the appropriate measures.
Our task is to provide all stakeholders with periodic information (such as this bi-monthly News Communiqué), market updates, material news flow, reassurance and comfort when due, but also negative news, if and when it emerges.
Finally, on a high note, the outlook for CLINUVEL is bright despite the global coronavirus pandemic. Our Board and management team are justifiably optimistic about the Company, its commercial future and growth.
SUMMARY
For those who have followed CLINUVEL for longer, there may be recognition in our pattern and approach to the business. Despite yesterday’s successes, we are seldom content and strive to maintain status quo or progress.
In the context of the global economic changes taking place, we project and plan all activities to further execute and build value. The challenges are enormous, both from the perspective of taking technology to a mature and commercial level but also in anticipating risks. CLINUVEL’s outlook is good, but it all hinges on our initiation, discipline, and hunger to execute. The platform to work from is as strong as we had desired in 2005. But past performance means very little in my view, we had a great year but there is very little reason to celebrate in today’s environment.
In the September News Communiqué V, we will speak more about the DNA program, vitiligo and the new indications awaiting Ethics and Regulatory approvals. Although we are frustrated by the current lengthy review by the various authorities, we are in full preparation to start when the paperwork is signed off.
My team has reported encouraging support of long-term owners for CLINUVEL’s performance, and on this occasion, I express my appreciation to you all.
Philippe Wolgen