CLINUVEL Kommuniqué 1
| Melbourne, Australia, 27 January 2022 |
ASX: XETRA-DAX: Level 1 ADR: |
CUV UR9 CLVLY |
Liebe Aktionäre, Freunde,Liebe Aktionäre, Freunde,
Einleitung
(Herr Malcolm Bull, Head of Australian Operations & Investor Relations)
Allen unseren Stakeholdern ein frohes neues Jahr für 2022, ein Jahr, in dem CLINUVEL Fortschritte bei unseren strategischen Initiativen in allen Geschäftsbereichen unseres Unternehmens erwartet. Zu Beginn des dritten Jahres der COVID-19-Pandemie fühlen wir uns von den weltweiten Fortschritten bei der Entwicklung und Verabreichung von Impfstoffen ermutigt. Wir fühlen mit allen Betroffenen der Pandemie und bewundern und schätzen das Engagement der Mitarbeiter im Gesundheitswesen der gesamten Welt. Während wir weiterhin die Auswirkungen auf unser Geschäft verwalten und die Verteilung von Auffrischimpfstoffen zur Bekämpfung der Delta- und Omicron-Varianten überwachen, ist das CLINUVEL-Team gestärkt, das Wachstum des kommerziellen Betriebs und die Erweiterung des klinischen Programms voranzutreiben.
Das Finanzmanagement von CLINUVEL bietet dem Unternehmen die Möglichkeit, externe Faktoren wie die Pandemie zu bewältigen. Unsere Ziele haben sich daher nicht geändert und CLINUVEL macht trotz des vorherrschenden Betriebsumfelds weiterhin Fortschritte beim Aufbau einer unabhängigen Unternehmensgruppe.
Dieses erste Nachrichtenkommuniqué von 2022 enthält ein Update zu:
- Handelsbetrieb;
- Peer-Reviews der Erfahrungen von EPP-Patienten mit SCENESSE®;
- linische Programme; und
- Kommunikation und Investor Relations.
Während wir weiterhin die Auswirkungen auf unser Geschäft verwalten und die Verteilung von Auffrischimpfstoffen zur Bekämpfung der Delta- und Omicron-Varianten überwachen, ist das CLINUVEL-Team gestärkt, das Wachstum des kommerziellen Betriebs und die Erweiterung des klinischen Programms voranzutreiben.
Behandlung von Patienten mit erythropoetischer Protoporphyrie (EPP) in den USA
(Dr. Linda Teng, Direktorin Nordamerika)
Spezialzentren und Versicherer
Über 40 spezialisierte EPP-Zentren wurden in den USA geschult und akkreditiert. Diese Zentren liegen strategisch günstig in der Nähe der Wohnhäuser von EPP-Patienten. Das US-Team identifiziert und schult weiterhin Zentren an neuen Standorten basierend auf dem Cluster bekannter EPP-Patienten. Seit Mitte 2021 ist ein Anstieg von 60 auf über 100 gewerbliche und staatliche Versicherer zu verzeichnen, die nun klinische Policendokumente für SCENESSE® veröffentlicht haben und die SCENESSE®-Behandlung im Rahmen einer vorherigen Genehmigung (PA) abdecken. Mit diesen klinischen Richtlinien, erhalten Patienten mit bestätigten EPP-Diagnosen PA-Zulassungen und die Abdeckung des Medikaments.
Darüber hinaus hat das Medicare-Programm damit begonnen, SCENESSE® für eine kleine Gruppe von EPP-Patienten abzudecken. Medicare, das staatliche Krankenversicherungsprogramm, das von den Centers for Medicare & Medicaid Services (CMS) verwaltet wird, bietet eine Krankenversicherung für Amerikaner, die 65 Jahre oder älter sind, bestimmte jüngere Menschen mit Behinderungen und Amerikaner mit Endstadium Nierenkrankheit.
Das SCENESSE®-Sparprogramm wurde für berechtigte EPP-Patienten im Rahmen von privaten und/oder gewerblichen Versicherungsplänen eingerichtet. Dieses Programm wurde erfolgreich umgesetzt und hat berechtigten EPP-Patienten seit Beginn des Marktzugangs im Jahr 2020 finanzielle Unterstützung gewährt.
Seit Mitte 2021 ist ein Anstieg von 60 auf über 100 gewerbliche und staatliche Versicherer zu verzeichnen, die nun klinische Policendokumente für SCENESSE® veröffentlicht haben und die SCENESSE®-Behandlung im Rahmen einer vorherigen Genehmigung (PA) abdecken.
Behandlungscodes, Implementierung und Auswirkungen
Behandlungscodes für die Abrechnung von SCENESSE®-Medikamenten und -Verfahren zu Versicherungsansprüchen sind nun vollständig festgestellt. Der Level-II-HCPCS-Medikamenten-J-Code (J7352) für SCENESSE® als „Afamelanotid-Implantat, 1 mg“ wurde im Januar 2021 vom HCPCS (Healthcare Common Procedure Coding System) vergeben. Der Verabreichungsverfahrenscode CPT 11981 „Insertion, Drug Delivery Implant (dh bioresorbable, biodegradable, nonbiodegradable)“ wurde von der AMA (American Ärztekammer) im Jahr 2021 mit Wirkung ab Januar 2022 ueberarbeitet.
In den meisten Fällen erfordert die Abdeckung von SCENESSE® (J7352) weiterhin eine PA-Genehmigung, während der aktualisierte Verfahrenscode für die SCENESSE®-Verwaltung (CPT 11981) keine PA-Genehmigung mehr erfordert. Diese Änderung wird die Bearbeitungszeit der Zentren verringern. Die Versicherer erstatten SCENESSE® angemessen gemäß dem zwischen einzelnen Zentren und Versicherern vereinbarten Satz J7352. Bei der Umsetzung der Änderungen in der Coderegistrierung unterstützen wir oder managen aktiv die Diskrepanzen, Fehler und Verzögerungen, die für Patienten entstehen, um eine Behandlung zu erhalten, aber insgesamt ist eine erste Bewertung so, dass der neue Abrechnungscode die Effizienz des Abrechnungsprozesses gleichermassen für Versicherer und die Zentren verbessert hat.
Patientennachfrage und Erfahrungen
Tatsächlich war das letzte Jahr (2021) das erste vollständige Kalenderjahr der SCENESSE®-Behandlung, und es wurde beobachtet, dass die Mehrheit der EPP-Patienten die Behandlung während der gesamten Wintersaison fortsetzte. Während CLINUVEL nach dem australischen Geschäftsjahr (1. Juli bis 30. Juni) berichtet, überwachen wir auch die Leistung des Kalenderjahres für regulatorische Zwecke. Wie in den Vorjahren begann eine höhere Nachfrage nach Behandlungen um die Frühjahrs- und Sommersaison herum. Wir werden abwarten, ob der gleiche Trend im kommenden Jahr zu beobachten ist, während unser Team weiterhin EPP-Patienten an Spezialzentren überweist und sie bei der PA-Zulassung unterstützt. Wir haben gesehen, dass in den letzten Wochen die Aktivität bei PA-Einreichungen und/oder Verlängerungen in Vorbereitung auf die Frühjahrssaison zugenommen hat.
Im vergangenen Jahr haben unsere Klinik- und Vertriebsteams positive Rückmeldungen und Anekdoten zur verbesserten Lebensqualität nach der Behandlung mit SCENESSE® erhalten. Diese realen Erfahrungen liefern uns wertvolle Erkenntnisse wie Patienten davon profitieren und wie sie ein Leben führen können, das zuvor für unvorstellbar gehalten wurde. Mit verstärkter Kraft und neuen Teammitgliedern freuen wir uns darauf, Spezialzentren und Patienten dabei zu unterstützen, sicherzustellen, dass sie weiterhin eine außergewöhnliche Versorgung und verbesserte Lebensqualität durch die SCENESSE®-Behandlung erhalten.
Wir haben einen konkreten Plan, an den wir uns halten, wenn es um die langfristige klinische Versorgung geht; dieser traegt in Europa und den USA bereits Früchte und geben den Aufsichtsbehörden viel Vertrauen, dass wir uns tatsächlich um unsere Patienten kümmern.
Behandlung von EPP-Patienten in Europa
(Herr Lachlan Hay, Director of Global Operations)
EPP-Patienten suchen weiterhin in ganz Europa nach einer Behandlung mit SCENESSE® (Afamelanotid 16 mg), trotz der anhaltenden Einschränkungen durch die COVID-19-Pandemie. Basierend auf ersten Rückmeldungen erwarten wir, dass einige europäische EPP-Expertenzentren im Jahr 2022 weiterhin mit begrenzter Kapazität arbeiten werden, da Ressourcen für die kritische Patientenversorgung umgeleitet und der Rückstand der Wahlversorgung aufgeholt werden muss. Die EPP-Expertenzentren sind sich jedoch bewusst über die Vorteile einer kontinuierlichen Versorgung von EPP-Patienten bewusst, anstatt sie aus der Behandlung zu nehmen (siehe Kommentar von Dr. Bilbao unten). EPP bleibt eine komplexe Krankheit, die eine multidisziplinäre Behandlung erfordert.
Fortführung von SCENESSE® in Deutschland
Wie Anfang dieser Woche angekündigt, wurde ein äußerst wichtiger Meilenstein erreicht, indem unser Team eine zweite Vereinbarung mit dem Deutschen Spitzenverband der Gesetzlichen Krankenkassen (GKV-SV) abgeschlossen hat. Diese Vereinbarung sichert deutschen EPP-Patienten den Zugang zu SCENESSE®. In Vorbereitung auf diese langwierigen, aber notwendigen Verhandlungen analysierte unser Team Vertriebs- und Patientendaten, überprüfte klinische Studiendaten und die Behandlungsansprechrate. Ein Team von Experten, Beratern und klinischen und kommerzielle Mitglieder kamen zu dem Schluss, dass die Position und der Ansatz von CLINUVEL zur Aushandlung eines fairen und angemessenen Preises gerechtfertigt waren. Gestärkt im Vertrauen darauf, dass CLINUVEL sein Versprechen gegenüber deutschen, aber auch anderen nationaler Versicherern gehalten hat, wurden die Gespräche mit dem Gemeinsamen Bundesausschuss (G-BA) und dem GKV-SV unter Ausschluss der Öffentlichkeit geführt.
Zwei öffentliche Sitzungen wurden abgehalten, als unsere Teams einen unfairen Versuch der Verhandlungsführer aufdeckten, andere unbegründete Daten zu präsentieren, aber schließlich während den Verhandlungen wurde anhand der geprüften Daten von CLINUVEL nachgewiesen, dass Unsere Position felsenfest war und auf Fakten und Daten basierte. Ich habe dieses positive Ergebnis in der öffentlichen Ankündigung von CLINUVEL (ASX: 24. Januar 2022) als direktes Ergebnis unserer Strategie und unseres gerechten Ansatzes in ganz Europa bezeichnet.
Es gibt viele EPP-Patienten und Familien in Deutschland, die seit Beginn unseres kommerziellen Programms im Jahr 2016 kontinuierlich in EPP-Expertenzentren betreut und behandelt wurden, wobei einige zuvor an unseren Programmen für klinische Studien und Compassionate Access teilgenommen haben. Alle diese Patienten liefern wertvolle Daten aus dem wirklichen Leben, die die bereits produzierten Daten aus klinischen Studien ergänzen.
…wurde ein äußerst wichtiger Meilenstein erreicht, indem unser Team eine zweite Vereinbarung mit dem Deutschen Spitzenverband der Gesetzlichen Krankenkassen (GKV-SV) abgeschlossen hat. Diese Vereinbarung sichert deutschen EPP-Patienten den Zugang zu SCENESSE®
Der Erstattungsprozess in Deutschland ist im Sozialgesetzbuch V (SGB V) gesetzlich vorgeschrieben und beinhaltet Interaktionen mit und zwischen drei unterschiedlichen, aber sich überschneidenden Einheiten:
- der G-BA;
- das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (iQWiG); und
- der GKV-SV
Eine weitere Rahmenvereinbarung – definiert unter § 130b des SGB V – ordnet die Prozesse und Interaktionen zwischen Arzneimittelzulassungsinhabern (MAHs) und der GKV-SV an, um Vereinbarungen über die Erstattung zu treffen, was die Fähigkeit von rund 90% der deutschen Bevölkerung Zugang zu einer Behandlung erhalten, betrifft. (Die anderen 10 % der Bevölkerung sind privat versichert durch Mitglieder der Privaten Krankenversicherung, PKV, die Beobachter des GKV-Prozesses sind.) Schließlich wird das Gesundheitsministerium auch an der Bewertung des Marktzugangs neuartiger Therapien beteiligt sein.
Von der ersten Interaktion bis zur endgültigen Vereinbarung können Gespräche und Preisverhandlungen 18 bis 24 Monate dauern, ein Weg, den alle Unternehmen gehen müssen, um den Marktzugang für Therapien in Deutschland zu ermöglichen oder aufrechtzuerhalten. In den ersten 12 Monaten nach der Einführung eines Produkts in Deutschland kann ein MAH den Marktpreis eines Produkts frei festlegen, kann jedoch in den Folgezeiträumen während des Abschlusses des GKV-SV-Verfahrens hohen Rückforderungen ausgesetzt sein.
Die ersten Interaktionen und Diskussionen in der deutschen Erstattung Prozessorientierung auf den „Nutzen“ eines Medikaments, unabhängig von den bereits durchgeführten Nutzenbewertungen der Europäischen Arzneimittelagentur (EMA).
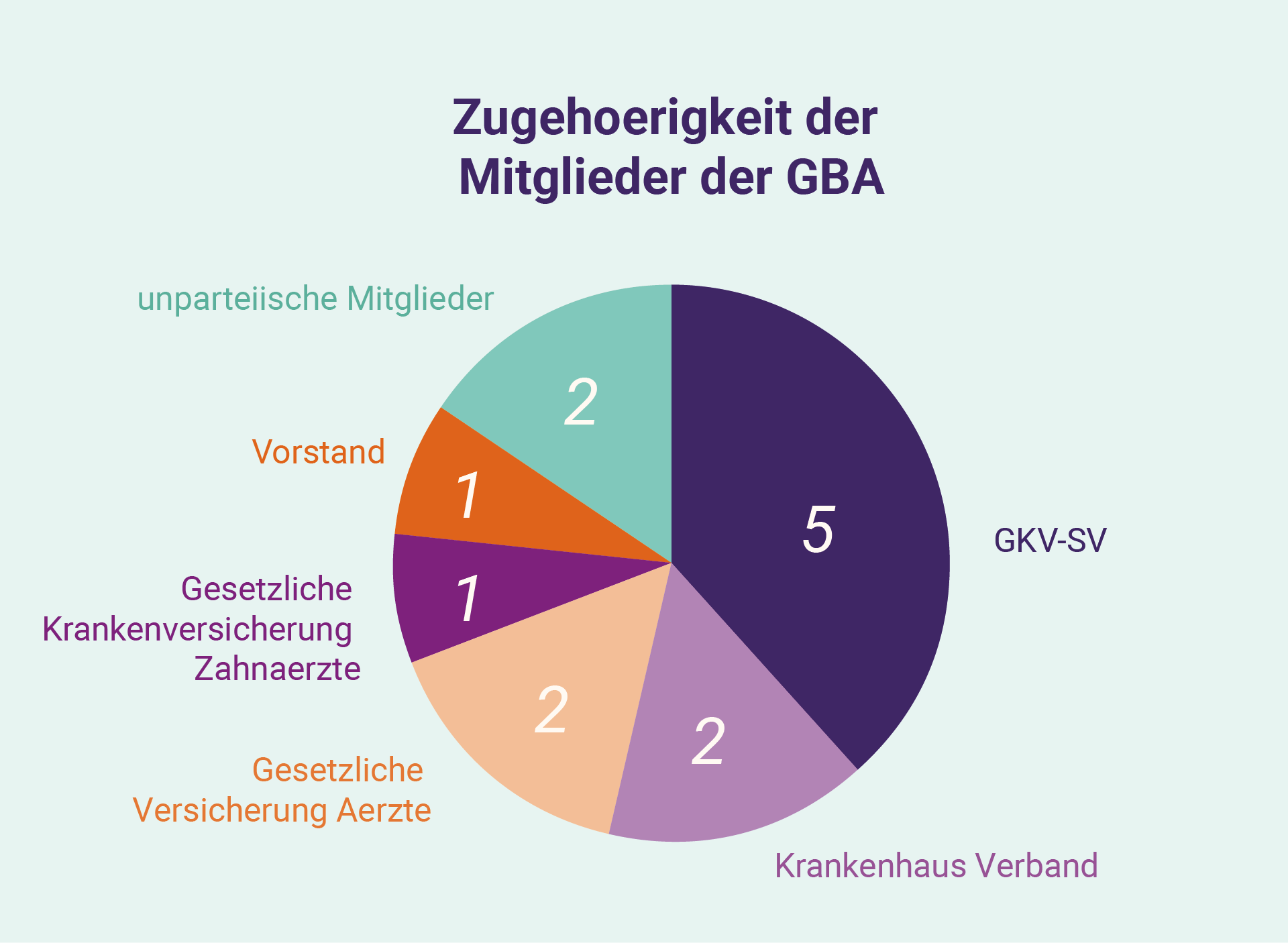
Der G-BA-Ansatz zum „Nutzen“
Die ersten Interaktionen und Diskussionen in der deutschen Erstattung Prozessorientierung auf den „Nutzen“ eines Medikaments, unabhängig von den bereits durchgeführten Nutzenbewertungen der Europäischen Arzneimittelagentur (EMA). Die Nutzenbewertung erfolgt durch den G-BA, ein 13-köpfiges Gremium bestehend aus: je zwei Vertretern der Krankenhausgesellschaft und Kassenärzten; ein Vertreter der Kassenzahnärzte; fünf Vertreter des GKV-SV; zwei unparteiische Mitglieder; und einen unparteiischen Vorsitzenden. § 35a des Das SGB V verlangt diese Bewertung des Vorteils (einschließlich gegen eine „angemessene Vergleichsperson“, ein „Zusatznutzen“ und die therapeutische Bedeutung) eines Arzneimittels durch den G-BA, wobei die Zulassungsinhaber zur Untermauerung ihrer Angaben Dossiers und Nachweise einreichen müssen. Unternehmen können den G-BA vor dieser Einreichung beauftragen, um zu verstehen, wie der Nutzen vom G-BA verstanden werden kann, und die eingereichten Nachweise zu präzisieren. Diese Dossiers – die mehrere hundert Seiten umfassen – beinhalten die Entwicklung und klinische Studien für ein Produkt in einer bestimmten Patientenpopulation, zusätzlich gesammelte Nachweise (z. B. Nachweise aus der Praxis) und umfangreiche Peer-Review-Literatur. Der G-BA kann als Antwort auf die Einreichung in den folgenden Monaten weitere Informationen oder Unterlagen einholen, da er zwei Dokumente zur Veröffentlichung vorbereitet: eine Nutzenbewertung („Nutzenbewertung“) und eine Begründung („Tragende Gründe“).
Bei einem Orphan Drug ist der „Nutzen“ des Produktes nach deutschem Recht bereits anerkannt (es sei denn, der Umsatz wird voraussichtlich eine definierte Schwelle überschreiten), und die Rolle des G-BA besteht in erster Linie darin, den Nutzen des Produktes , Kategorisierung nach niedrig-mittel-hoch oder „nicht quantifizierbar“ in der Tragende Gründen festzulegen. Diese Kategorisierung wirkt sich unmittelbar auf die späteren Interaktionen mit GKV-SV aus.
Der G-BA ist hinsichtlich der Annahme oder Zurückweisung von Nachweisen nicht eingeschränkt bei der Bewertung bis auf die Ebene einzelner Endpunkte in konkreten klinischen Studien. Diese werden nach ihren Auswirkungen auf Mortalität, Morbidität, Lebensqualität und Sicherheit der Patienten kategorisiert. Während der Begutachtung kann das iQWiG vom G-BA beauftragt werden, unabhängige Bewertungen eines vollständigen Dossiers oder bestimmter Elemente eines Dossiers gemäß den vom G-BA definierten Bedürfnissen durchzuführen, einschließlich bestimmter Nachweise oder Endpunkte oder breiterer Informationen (z die Größe einer Patientenpopulation). Die Nutzenbewertung skizziert die Gründe für die Akzeptanz oder Ablehnung von Evidenz, einschließlich aller Endpunkte, die auf der Grundlage ihrer Operationalisierung, Patientenrelevanz und Gueltigkeit identifiziert wurden. Weitere Bewertungen beinhalten, ob in anderen Regionen erhobene Evidenz auf die deutsche Situation übertragbar ist und ob der G-BA bestimmte Evidenz oder Studien als verzerrt ansieht.
Ungefähr vier Monate nach Einreichung des Dossiers wird ein Entwurf der Nutzenbewertung veröffentlicht und ist offen für Rückmeldungen des Inhabers der Genehmigung für das Inverkehrbringen der auch eine mündliche Anhörung zur Erörterung der Berichte, Beweismittel und Entscheidungen des G-BA beantragen kann. Der MAH kann in der mündlichen Anhörung erneut seinen Standpunkt darlegen und versuchen, auf bestimmte vom G-BA (bzw. iQWiG) angesprochene Punkte in den Berichten einzugehen Anschließend wird eine abschließende Nutzenbewertung erstellt und veröffentlicht.
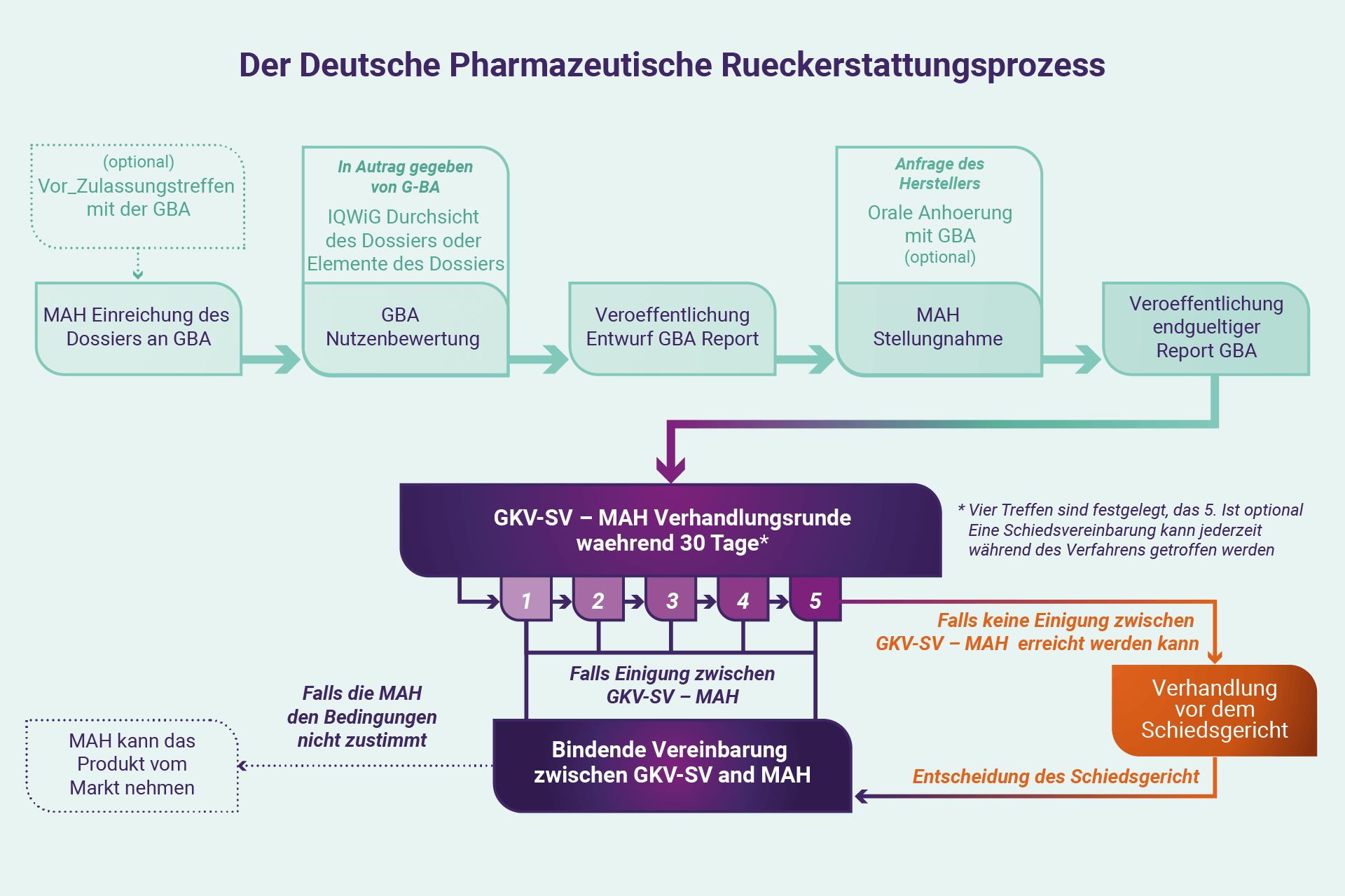
Vom G-BA zum GKV-SV
Der Erstattungsprozess verlagert sich dann von Interaktionen mit dem G-BA und GKV-SV. Die GKV-SV nutzt im Rahmen der Rahmenvereinbarung die Nutzenbewertung als Grundlage, um den Nutzen einer Therapie im Verhandlungsprozess zu diskutieren: eine Serie von vier Sitzungen (mit einer optionalen fünften) von jeweils vier Stunden (in Wirklichkeit meist viel mehr) , wo Vertreter des GKV-SV und MAH werden gebeten, eine Einigung zur Ermöglichung zu erzielen oder weiterhin Zugang zu einer Therapie. Verhandlungsgespräche – grob abgehalten im Abstand von einem Monat – konzentrieren sich zunächst auf die Krankheit und dann auf den Nutzen der Behandlung in Preisgestaltung, Erstattung (im Allgemeinen mit Fokus auf erwartete Rabatte), und allgemeine Auswirkungen auf den nationalen und regionalen Haushalt. Von besonderer Relevanz sind die Preise für eine Therapie eines Herstellers in anderen Ländern, die Höhe der angebotenen Diskontierung und die Kosten von Vergleichstherapien für die Krankheit. Generell erwartet der GKV-SV von den Herstellern einen Rabatt über die normalerweise vorgeschriebenen sieben Prozent um den Eintritt in den deutschen Markt zu ermöglichen. Die Verhandlungen werden vertraulich behandelt wie es die gesetzlichen Verpflichtungen vorschreiben.
Die Sitzungen des GKV-SV finden jeweils im Plenum (quasi in der heutigen Zeit) unter Vorsitz des GKV-SV mit Protokollführung statt. Außerhalb der formellen Treffen kann sich jede Partei auch an die andere wenden, um eine Einigung zu erzielen, oder alternativ erklären, dass sie die Differenzen für unüberbrückbar halten und die Intervention eines Schiedsgerichtshofs beantragen, dessen Entscheidungen letztendlich für beide Parteien bindend sind. Akzeptiert ein Hersteller die Feststellungen des Gerichts nicht, kann er sich letztendlich sein Produkt vom deutschen Markt zurückziehen.
Daher kann jedes Jahr jeweils eine umfangreiche Liste verschreibungspflichtiger Medikamente vom deutschen Markt genommen warden. Wenn im Laufe des Verfahrens jedoch eine Einigung erzielt werden kann, zwischen beiden Parteien, kann ein förmlicher Vertrag unterzeichnet werden.
Ein erfolgreiches Ergebnis
Zum zweiten Mal in sechs Jahren hat das Team von CLINUVEL diesen Prozess erfolgreich abgeschlossen, wobei die Neubewertung durch den G-BA im Jahr 2021 durch eine zeitliche Begrenzung ihrer ursprünglichen Entscheidung im Jahr 2016 ausgelöst wurde. 2017 wurden die Vorteile von SCENESSE® und sein fairer klinischer Wert vom Healthcare Court of Arbitration anerkannt, um deutschen Patienten den notwendigen Behandlungszugang zu ermöglichen. In Übereinstimmung mit unseren öffentlichen Erklärungen und nachgewiesen durch jährliche Finanzprüfungen hat das Unternehmen eine transparente, einheitliche Preispolitik eingeführt und bietet keine Rabatte, Preisnachlässe oder Cashbacks an, die am häufigsten und häufig in Preisdiskussionen mit staatlichen und privaten Versicherern erwartet werden; der einzige Mechanismus, der Unternehmen zur Verfügung steht, um den Patientenzugang zu ermöglichen.
In unseren Interaktionen seit 2017 und insbesondere im Laufe des Jahres 2021 haben deutsche Versicherer viel Vertrauen in die Policen von CLINUVEL gewonnen und kommen zumVerstaendniss, wie auch sie von einem solchen gerechten Ansatz profitieren. Während die Details wie bei allen GKV-SV-Vereinbarungen vertraulich zu behandeln sind, Das Endergebnis ist, dass EPP-Patienten in Deutschland fortfahren können Behandlung zu erhalten. Es muss auch als breiterer Sieg für die EPP Patienten in ganz Europa angesehen werden, wobei Deutschland als ein wichtiges Referenzland für getroffene Erstattungsentscheidungen ist.
Während wir mit dem Ergebnis des GKV-SV zufrieden und bescheiden bleiben, schätzen wir weiterhin das Engagement der GKV-SV-Führung. Das Team war regelmäßig frustriert über die Art und Weise, wie Patienten behandelt wurden und Daten wurden von Vertretern einiger Stellen, die für die Nutzenbewertung zuständig sind, falsch dargestellt oder ignoriert, was letztendlich dazu führt, dass EPP-Patienten den Zugang zu der einzigen für ihre Erkrankung zugelassenen Behandlung erschweren. Letztendlich werden die Versicherer die Wirtschaftlichkeit prüfen und Mittel anderen Behandlungen zuweisen oder wo Rabatte gezahlt werden.
In unseren Interaktionen seit 2017 und insbesondere im Laufe des Jahres 2021 haben deutsche Versicherer viel Vertrauen in die Policen von CLINUVEL gewonnen und kommen zumVerstaendniss, wie auch sie von einem solchen gerechten Ansatz profitieren.
SCENESSE® wurde 2014 von der EMA zugelassen. Bei der Zulassung des Produkts, erkannte die Agentur – mit der Überprüfung des Registrierungsdossiers unter der Leitung von Berichterstattern aus die deutsche Agentur BfArM – der Einzigartigkeit von EPP als Stoffwechselstörung und die Herausforderungen bei der Quantifizierung der Auswirkungen der Erkrankung auf einen Patienten (und damit die Wirkung einer Therapie). Dies wurde im Europäischen Öffentlichen Bewertungsbericht für SCENESSE® zusammengefasst:
„Unter normalen Anwendungsbedingungen erlaubt der Stand der aktuellen wissenschaftlichen Erkenntnisse, Werkzeuge und Instrumente keine ausreichend präzisen Messungen der Auswirkungen von Krankheiten durch‚ sichtbares Licht‘ auf exponierte Haut.“
Die EMA erkannte auch an, dass ein weiteres Vorgehen unethisch wäre, klinische Studien mit Afamelanotid bei EPP-Patienten, einer Patientenpopulation, die möglicherweise nicht in der Lage oder willens, an weiteren Studien teilzunehmen, aufgrund der Komplexität und des Risikos, sich Verbrennungen 3. Grades zuzuziehen, während sie von absoluter Lichtunverträglichkeit leiden. Anders gesagt, die einzigartigen Herausforderungen der EPP verlangen, dass bei der Beurteilung des Behandlungsnutzens andere Ansätze als die „üblichen“ Bewertungswege in Betracht gezogen werden. Wie belegt durch die US Food and Drug Administration – die die reale Welt einbezog; Beweise in ihre positive Nutzen-Risiko-Bewertung von SCENESSE® für EPP – solche Ansätze werden zumindest zunehmend als Weg nach vorn bei der Bewertung neuartiger Therapien anerkannt. In vielerlei Hinsicht war SCENESSE® ein Maßstab für die EMA und die FDA, auf den sie sich für andere medizinische Produkte fuer seltene Krankheiten beziehen konnten.
Peer Reviews der Erfahrungen von EPP-Patienten mit SCENESSE®
(Dr Pilar Bilbao, Leiterin des klinischen Betriebs)
Während wir die Nachzulassungs- und Sonderzugangsprogramme in Europa vorangetrieben haben, hat unser Team mehr über EPP und seine Behandlung unter realen Bedingungen gelernt, wertvolle Erkenntnisse hinzugefügt, Bewertungsinstrumente validiert und – vor allem – eine genaue (oft tägliche) Überwachung des Sicherheitsprofil des Produkts durchgefuehrt. Die Datenmenge zur Verwendung des Produkts in EPP wächst weiter und gibt uns Sicherheit hinsichtlich des langfristigen Nutzen-Risiko-Profils von Afamelanotid.
Zwei kürzlich veröffentlichte Peer-Review-Artikel von europäischen Expertenzentren veranschaulichen dies, von denen einer spannende Einblicke in eine bisher nicht berichtete Wirkung bei EPP-Patienten bietet: Schutz vor Leberschäden. Während unser Team eine Ahnung von den möglichen systemischen und hepatischen Wirkungen von Afamelanotid hatte 16 mg in Form mit kontrollierter Freisetzung, war es die Schweizer Gruppe, der zugeschrieben wird, Beweise für einen Leberschutz bei Langzeitanwendung gefunden zu haben.
Etwa bis zu 20 Prozent der EPP-Patienten erleiden im Laufe ihres Lebens irgendeine Art von Leberschädigung als Folge der Akkumulation und Speicherung von Protoporphyrin IX – PPIX; das gleiche Vorläufer-Porphyrin, das für die Phototoxizität verantwortlich ist. Bei etwa 2 bis 5 Prozent der EPP-Patienten schreitet diese Schädigung zu einem Leberversagen fort, was eine lebensrettende Lebertransplantation und eine anschließende lebenslange Immunsuppression erforderlich macht. Unsere Teams konzentrieren sich seit langem auf die medizinischen Bedingungen, die sich aus der Immunsuppression ergeben, ein Wissen, das wir in anderen Programmen wie den aktuellen DNA-Reparaturstudien verwenden. Dieses schwere EPP-Symptom der Hepatotoxizität erfordert jedoch, dass die Leberfunktion von EPP-Patienten regelmäßig überprüft wird und dass die Patienten Vorsichtsmaßnahmen ergreifen, um Leberschäden zu vermeiden, indem sie bestimmte pharmazeutische Produkte sowie Alkohol meiden.
Zwei kürzlich veröffentlichte Peer-Review-Artikel von europäischen Expertenzentren veranschaulichen dies, von denen einer spannende Einblicke in eine bisher nicht berichtete Wirkung bei EPP-Patienten bietet: Schutz vor Leberschäden.
Leberschäden können mit einem Bluttest beurteilt werden, der spezifische Enzyme und Proteine exprimiert. Insbesondere bei EPP-Patienten werden PPIX-Spiegel gemessen, wobei erhöhte Werte darauf hindeuten, dass die Leber möglicherweise unter Belastung steht, um Giftstoffe aus dem Blutkreislauf zu filtern.
EPP-Patienten in der Schweiz werden bereits seit 2006 in verschiedenen Programmen mit Afamelanotid behandelt, wobei die erste klinische Studie überhaupt am Triemli-Spital in Zürich durchgeführt wurde (anschliessend im New England Journal of Medicine veröffentlicht). In einer über zwei Jahre durchgeführten Studie1 wurden spezifische Laborwerte bei 38 EPP-Patienten bewertet, die zu dem Schluss kamen, dass die Behandlung mit Afamelanotid die Konzentrationen sowohl von PPIX als auch von Aspartataminotransferase (AST), einem Enzym, das von der Leber als Reaktion auf eine Schädigung produziert werden kann, signifikant reduzierte. Am deutlichsten war der Rückgang bei den Patienten mit höheren Anfangsspiegeln sowohl von PPIX als auch von AST, wobei die Autoren diese Verringerung des Gesamtrisikos der Patienten für Leberschäden vorschlagen. Zink-Protoporphyrin (ZnP) nahm während der Behandlung ebenfalls ab. ZnP wird in roten Blutkörperchen gefunden, wenn die Hämproduktion durch Ursachen wie Eisenmangel gehemmt ist. Die Autoren erklärten dies mit einer verbesserten Verwertung von systemisch verfügbarem Eisen.
Diese beiden Erkenntnisse sind wichtige Entwicklungen, die zu unserem breiteren Verständnis der Mechanismen beitragen, durch die Afamelanotid möglicherweise abgebaut wird in der Lage, EPP – und vielleicht auch anderen – Patienten langfristig zu helfen. Wenn gezeigt werden kann, dass die Behandlung mit Afamelanotid neben dem systemischen Lichtschutz auch einen Leberschutz bietet, würde dies die Krankheitslast noch weiter verringern und einen Rundumschutz bei dieser seltenen Stoffwechselstörung bieten. Eine zweite Studie ist im Gange, um diese ersten veröffentlichten Ergebnisse und folgende Schlussfolgerungen zu bestätigen; Wir werden dann den am besten geeigneten Weg nach vorne bewerten.
Diese beiden Erkenntnisse sind wichtige Entwicklungen, die zu unserem breiteren Verständnis der Mechanismen beitragen, durch die Afamelanotid möglicherweise abgebaut wird in der Lage, EPP – und vielleicht auch anderen – Patienten langfristig zu helfen.
Das zweite Papier2 des niederländischen EPP-Expertenreferenzzentrums hat sich mit den Auswirkungen einer Behandlung mit Afamelanotid auf den zirkadianen Rhythmus (ein natürlicher, interner Prozess, der den Schlaf-Wach-Zyklus reguliert) und die allgemeine Lichtexposition befasst und eine Kohorte von EPP-Patienten, die sich einer Behandlung unterziehen, abgeglichen zu sowohl unbehandelte Patienten als auch eine Kontrollgruppe. Im Laufe des Studiums wurde festgestellt, dass die Behandlung mit Afamelanotid es den Patienten ermöglichte, sich im Frühling mehr „weißem Licht“ auszusetzen, was zu weniger Schmerzen führte und die dazu beitrugen, den zirkadianen Rhythmus der Patienten zu normalisieren. Als Störungen des zirkadianen Rhythmus kann sich negativ auf die psychische Gesundheit, den Schlaf, und körperlicher Aktivität kann die Normalisierung des zirkadianen Rhythmus als Beitrag zu einem besseren allgemeinen Gesundheitszustand bei behandelten EPP-Patienten angesehen werden.
Klinische Programme
(Dr Pilar Bilbao, Leiterin des klinischen Betriebs)
Nachfolgend finden Sie einen Überblick über die wichtigsten Entwicklungen im erweiterten klinischen Programm von CLINUVEL:
Arterieller ischämischer Schlaganfall
Anfang dieses Monats gaben wir den Abschluss der Patientenrekrutierung für die CUV801-Studie bekannt, die weltweit erste Studie zur Sicherheit und Wirksamkeit von Afamelanotid zur Behandlung von Patienten mit arteriellem ischämischem Schlaganfall (AIS). Die Behandlung mit Afamelanotid wurde von allen sechs Patienten gut vertragen, und es wurden keine unerwünschten Arzneimittelwirkungen berichtet. Die Studie wird nun abgeschlossen und die Ergebnisse im Laufe des Jahres 2022 analysiert und geteilt. Die nächsten Studien werden bereits vorbereitet, wobei die Erkenntnisse aus den klinischen Beobachtungen und die Erkenntnisse aus CUV801 berücksichtigt werden.
Angesichts von schätzungsweise 15 Millionen Schlaganfällen, die jedes Jahr weltweit erlitten werden, kann man davon ausgehen, dass der größte Teil der Bevölkerung – direkt oder indirekt – von einem Schlaganfall betroffen ist. Viele Schlaganfallpatienten haben bleibende Hirnschäden und Behinderungen für das Leben, Verlust ihrer Fähigkeit zu funktionieren oder für sich selbst zu sorgen ohne fremde Hilfe, und es besteht ein geschätztes Risiko von 40 %, dass Patienten davon betroffen zu sein, einen Schlaganfall im folgenden Lebensjahrzehnt (11 % innerhalb des ersten Jahres) zu bekommen.3 Die Belastung durch diesen akuten Zustand für diejenigen Patienten, die überleben ist immens und fällt im Allgemeinen auf unmittelbare Familienmitglieder, die müssen oft ihr ganzes Leben grundlegend umstrukturieren, um die notwendige Pflege zu leisten.
Die Studie wird nun abgeschlossen und die Ergebnisse im Laufe des Jahres 2022 analysiert und geteilt. Die nächsten Studien werden bereits vorbereitet, wobei die Erkenntnisse aus den klinischen Beobachtungen und die Erkenntnisse aus CUV801 berücksichtigt werden.
Etwa 85 % aller Schlaganfälle sind ischämische Schlaganfälle, die Folge von Blutgerinnsel, das sich in einer Gehirnarterie festgesetzt hat und den Zellen Sauerstoff und Nährstoffe entzieht. Aufgrund von Zugang zur Behandlung und enge „therapeutische Zeitfenster“, in denen eine Behandlung verabreicht werden kann,gelten eine beträchtliche Anzahl dieser Patienten (bis zu 85 % in einer europäischen Studie4) als nicht geeignet für die Behandlungsstandards für pharmazeutische (Thrombolyse) oder chirurgische Eingriffe (Thrombektomie). Einfach gesagt: Ärzten fehlen weitere notwendige Werkzeuge zur Bekämpfung von Schlaganfällen und zur Genesung von Patienten.
Aufgrund dieses ungedeckten Bedarfs und der Beweise, die die Anwendung von Afamelanotid bei Schlaganfällen unterstützen, haben wir dieses wichtige und aufregende klinische Programm gestartet.
Als allererste Studie zu Afamelanotid bei Schlaganfallpatienten bleibt die Sicherheit Schwerpunkt von CUV801. Hier überwachen unsere Teams die wichtigsten Gesundheitsergebnisse für Patienten vor, während und nach der Behandlung genau, um die Auswirkungen zu verstehen wie die Therapie sich auf ihren allgemeinen Gesundheitszustand auswirken kann. Unerwünschte Wirkungen, die auf die interventionelle medikamentöse Behandlung zurückzuführen sein können, sind hier ein entscheidender Faktor. Die Tatsache, dass wir bisher keine Nebenwirkungen im Zusammenhang mit der Behandlung mit Afamelanotid in der Studie gehabt hatten, gibt uns viel Trost, dass das Schlaganfallprogramm fortgesetzt werden kann. Wir warten nun auf den Abschluss der Studie durch die letzten Patienten und formale Datenanalysen, um die Auswirkungen von Afamelanotid in dieser Pilotstudie besser zu verstehen. Man darf den Fokus auf die Sicherheit bei der Verwendung unserer Melanocortine nicht unterschätzen, was für das Unternehmen in den kommenden Jahren einen großen Unterschied machen würde.
Um mehr über die Wissenschaft hinter der arteriellen Ischämie von CLINUVEL zu erfahren Schlaganfallprogramm, siehe:
DNA-Reparatur
Das DNA-Reparaturprogramm zur Bewertung der Schutzfunktion von Afamelanotid Haut und Regeneration der DNA der lichtgeschädigten Haut ist im Gange. Der Fokus liegt zunächst auf Patienten mit Xeroderma pigmentosum (XP). die 10.000-mal anfälliger für Hautkrebs sind und die im Laufe ihres Lebens an einer großen Anzahl dieser Krebsarten leiden, die oft zu Operationen und zum Tod führen. Zwei XP-C-Patienten wurden waehrend der CUV156-Studie behandelt. Eine Studie an gesunden Freiwilligen (CUV151) und weitere Studien mit XP-C- und XP-V-Varianten der Krankheit sind geplant.Ueber Fortschritt in diesen Studien wird im Laufe des Jahres 2022 berichtet.
Vitiligo
Vor Ende des Jahres 2021 gab CLINUVEL bekannt, dass es mit der Bewertung der Rolle von SCENESSE® als Monotherapie für die Behandlung der Depigmentierungsstörung der Haut, Vitiligo fortfahren wird. Dies kulminierte eine Periode der Verbindung mit der US Food and Drug Administration (FDA) über das Design der nächsten CUV-Studie. Da schmalbandige UV-B-Phototherapie immer noch nicht von der FDA für die Behandlung von Vitiligo zugelassen ist, stimmten wir zu, mit der Bewertung von SCENESSE® als Monotherapie zur systemischen Repigmentierung fortzufahren, die sich auf Patienten mit dunkleren Hauttypen (IV bis VI auf der Fitzpatrick-Skala) konzentriert. Wir arbeiten jetzt auf den Beginn der Pilotstudie CUV104 hin.
Eine Studie an gesunden Freiwilligen (CUV151) ist geplant, da sind weitere Studien mit XP-C- und XP-V-Varianten der Krankheit. Fortschritt in diesen Studien wird im Laufe des Jahres 2022 berichtet.
Andere Programme
CLINUVEL konzentriert sich weiterhin auf andere Programme, einschließlich einer Formulierung von Afamelanotid für pädiatrische EPP-Patienten, eine Studie zu SCENESSE® als Behandlung für Variegata Porphyrie (VP), eine Indikation im Zusammenhang mit EPP und eine neue (sechste) Indikation, die wir alle in diesem Jahr vorantreiben wollen.
Kommunikation und Investor Relations
(Herr Malcolm Bull, Head of Australian Operations & Investor Relations)
Die Häufigkeit und Reichweite der Kommunikation mit den Interessengruppen wird im Jahr 2022 noch weiter steigen. Dies spiegelt eine Zunahme der Aktualisierungen wider, die im gesamten erweiterten klinischen Programm wahrscheinlich sind. Die im Jahr 2021 abgehaltenen Betriebsaktualisierungen, strategischen Aktualisierungen und Investoren-Webinare wurden von der überwältigenden Mehrheit der Interessengruppen gut aufgenommen und werden auch 2022 fortgeführt.
Wir sind bestrebt, regelmäßig Informationen über den Fortschritt des Unternehmens zum Nutzen aller Interessengruppen und insbesondere für Investoren bereitzustellen, um weiterhin die positiven Gründe für ihre Investition in dieses dynamische Unternehmen mitzuteilen.
Der Jahresplan sieht regelmäßige Präsentationen auf Konferenzen, wie unten beschrieben:
| Monat | Veranstaltung |
|---|---|
| Januar | JP Morgan Healthcare Conference, San Francisco HC Wainwright Bioconnect-Konferenz |
| April | H.C. Wainwright Global Life Sciences-Konferenz |
| Mai | UBS Global Healthcare Conference |
| Juni | Jefferies Healthcare Conference |
| September | H.C. Wainwright Global Investment Conference |
| Oktober | Morgans 7th Annual Value in the Vines Investor Conference |
| November | Jefferies Gesundheitskonferenz in London |
2022 haben wir an der H.C. Wainwright Bioconnect Conference und an der JP Morgan Healthcare Conference teilgenommen. Beide fanden Anfang dieses Monats statt. Über 550 und 650 Unternehmen präsentierten sich auf jeder Konferenz. Aus den Konferenzen wurde ein allgemein positiver Ausblick auf 2022 gewonnen und das Feedback zu CUV war hervorragend. Der vermittelte Ansatz bestand darin, „damit weiterzumachen“, um die Coronavirus-Pandemie anzugehen und auf Unternehmensebene ihre Operationen und/oder klinischen Programme voranzutreiben. Dies spiegelt den Ansatz von CLINUVEL wider.
In Bezug auf bevorstehende Aktualisierungen werden wir in Kürze die Bareinnahmen und -ausgaben für das Dezemberquartal 2021 (das zweite Quartal des Geschäftsjahres 2022) bekannt geben, und Ende Februar werden die Finanzkonten für das Halbjahr bis Dezember 2021 veröffentlicht. Es wird ein Corporate Briefing geben zu den Cash-Ergebnissen und ein Investoren-Webinar zu den Halbjahresergebnissen. Auch Ankündigungen zum Fortschritt des klinischen Programms sind zu erwarten. Vorerst veröffentlichen wir aber weiterhin unsere Quartalsergebnisse das Unternehmen ist dazu nicht mehr verpflichtet, und ich sehe dies als Gefälligkeit an an unsere langjährigen unterstützenden Aktionäre.
In Bezug auf bevorstehende Aktualisierungen werden wir in Kürze die Bareinnahmen und -ausgaben für das Dezemberquartal 2021 (das zweite Quartal des Geschäftsjahres 2022) bekannt geben, und Ende Februar werden die Finanzkonten für das Halbjahr bis Dezember 2021 veröffentlicht.
Zusammenfassung und Schlussfolgerung
Es besteht kein Zweifel, dass 2022 ein ereignisreiches Jahr für CLINUVEL wie auch für uns wird. Weiterer Wachstum, zu einem vertikal integrierten und diversifizierten biopharmazeutischen Unternehmen expandieren, Fortschritte bei der erweiterten Palette klinischer Programme und beim anhaltenden Wachstum der kommerziellen Aktivitäten sind zu erwarten. Der Fokus des engagierten CLINUVEL-Teams wird mit umsichtigem Risiko- und Kostenmanagement aufrechterhalten um sicherzustellen, dass das Unternehmen seine Schlüsselinitiativen mit einem Leistungsergebnis vorantreibt, das im vorherrschenden Umfeld so stark wie möglich ist.
Wir wünschen allen Beteiligten ein sicheres und produktives Jahr 2022.
Malcolm Bull, Leiter des australischen Betriebs und der Investor Relations
References:
- Minder, et. al., (2022). Beyond pigmentation: signs of liver protection during afamelanotide treatment in Swiss patients with erythropoietic protoporphyria, an observational study. Therap Ad Rare Dis. 2: 1–17.
- Wensink, et. al., (2022). Objective light exposure measurements and circadian rhythm in patients with erythropoietic protoporphyria: a case-control study, Mol Genet and Metab. ePub 2 January 2022.
- Mohan, et. al., (2011). Risk and Cumulative Risk of Stroke Recurrence. Stroke. 42(5): 1489–94.
- de Sousa et. al., (2019). Access to and delivery of acute ischaemic stroke treatments: A survey of national scientific societies and stroke experts in 44 European countries. Eur Stoke J. 4(1):13–28.
Autorisiert für ASX-Freigabe durch den Verwaltungsrat von CLINUVEL PHARMACEUTICALS LTD.
Zukunftsgerichtete Aussagen
Diese Pressemitteilung enthält zukunftsgerichtete Aussagen, die die aktuellen Ansichten und Erwartungen des Managements von CLINUVEL widerspiegeln. Aussagen können eine Reihe bekannter und unbekannter Risiken beinhalten, die dazu führen können, dass unsere zukünftigen Ergebnisse, Leistungen oder Errungenschaften erheblich von denen abweichen, die in solchen zukunftsgerichteten Aussagen ausgedrückt oder impliziert werden. Wichtige Faktoren, die solche Unterschiede verursachen oder dazu beitragen könnten, umfassen Risiken in Bezug auf: unsere Fähigkeit, pharmazeutische Produkte zu entwickeln und zu vermarkten; die COVID-19-Pandemie und/oder andere weltweite, regionale oder nationale Ereignisse, die die Lieferkette über einen längeren Zeitraum betreffen, einschließlich unserer Fähigkeit, biopharmazeutische Produkte zu entwickeln, herzustellen, zu vermarkten und zu verkaufen; Konkurrenz für unsere Produkte, insbesondere SCENESSE® (afamelanotid 16mg), PRÉNUMBRA® oder NEURACTHEL®; unsere Fähigkeit, durch unsere innovativen F&E-Bemühungen die erwarteten Sicherheits- und Wirksamkeitsergebnisse zeitnah zu erzielen; die Wirksamkeit unserer Patente und anderer Schutzmaßnahmen für innovative Produkte, insbesondere angesichts nationaler und regionaler Unterschiede im Patentrecht; unsere potenzielle Gefährdung durch Produkthaftungsansprüche, soweit nicht durch eine Versicherung gedeckt; verstärkte staatliche Kontrolle unserer Vereinbarungen mit Dritten und Lieferanten in Australien, den USA, Europa, Israel, China und Japan; unsere Exposition gegenüber Währungsschwankungen und -beschränkungen sowie Kreditrisiken; die Auswirkungen von Reformen der Gesundheitsregulierung und der Arzneimittelpreise und -erstattung; dass das Unternehmen unerwartete Verzögerungen bei der ausgelagerten Herstellung von SCENESSE®, PRÉNUMBRA® oder NEURACTHEL® erleiden kann, was dazu führen kann, dass es seine kommerziellen Märkte und/oder klinischen Studienprogramme nicht beliefern kann; jegliche Nichteinhaltung von Melde- und Zahlungsverpflichtungen gegenüber staatlichen Zahlungssystemen (d. h. Medicare); Unsicherheiten im Zusammenhang mit den gesetzlichen und regulatorischen Pfaden für die Registrierung und Zulassung von Biotechnologie und verbraucherbasierten Produkten; Entscheidungen von Aufsichtsbehörden über die Zulassung unserer Produkte sowie deren Entscheidungen in Bezug auf Etikettenaussagen; unsere Fähigkeit, Schlüsselpersonal und Führungstalente zu halten oder anzuziehen; die Auswirkungen umfassenderer Veränderungen innerhalb der pharmazeutischen Industrie und verwandter Industrien; potenzielle Änderungen der Steuerverbindlichkeiten oder der Gesetzgebung; Umweltrisiken; und andere Faktoren, die in unserem Geschäftsbericht 2021 erörtert wurden.
Zukunftsgerichtete Aussagen beziehen sich nur auf das Datum, an dem sie gemacht werden, und das Unternehmen übernimmt keine Verpflichtung, abgesehen von den nach geltendem Recht oder den einschlägigen Notierungsvorschriften der Australian Securities Exchange vorgeschriebenen, zukunftsgerichteten Aussagen zu aktualisieren oder zu überarbeiten, sei es aufgrund neuer Informationen, zukünftiger Ereignisse oder aus anderen Gründen. Weitere Informationen zu vorläufigen und unsicheren Prognosen und Schätzungen sind auf Anfrage erhältlich, wobei darauf hingewiesen wird, dass die Wertentwicklung der Vergangenheit kein Indikator für die zukünftige Wertentwicklung ist.
Kontact
Level 11, 535 Bourke St
Melbourne, 3000 Vic,
Australia
+61 3 9660 4900
+61 3 9660 4909