SCIENTIFIC COMMUNIQUÉ VII
The Cerebral Vasculature System in Disease: Atherosclerosis
Introduction
The human brain is the control centre of the body and without it we could not survive. Predominantly comprised of neurons and glia, these cells collectively help form comprehensive communication circuits that enable our thoughts, memory, movement, and emotions. Owed to its constant activity, it is of no surprise the brain consumes approximately 20% of the body’s total oxygen supply under its physiological state alone. This organ is thus dependent on an efficient network of blood vessels to unceasingly deliver sufficient oxygenated blood to meets its high metabolic needs. As the brain coordinates almost all our functions, widespread impairment across the body can be seen in the absence of adequate blood flow to the brain. Mild chronic cerebral hypoperfusion has been linked to numerous neurodegenerative diseases, including Alzheimer’s disease. However, the sudden interruption of cerebral blood flow in an acute setting underpins one of the world’s most deadly diseases: stroke. Global figures suggest two thirds of stroke cases result in either permanent disability or mortality, with a stroke occurring every 40 seconds in the US. Secondary to the impact on the lives of stroke patients, this disease presents a sizeable economic burden, with current annual costs of stroke exceeding £25.6 billion and $45 billion in the UK and USA, respectively.
Haemorrhagic & Ischaemic Strokes
Strokes can be subcategorised based on their aetiology, which cause differing clinical manifestations within the brain. Broadly, they can be defined as either haemorrhagic or ischaemic, with the latter responsible for an estimated 85 to 87% of all recorded cases. Haemorrhagic strokes result from the rupturing of a weakened blood vessel directly within the brain, or alternatively within the brain’s subarachnoid space. This leads to the accumulation of blood within the brain tissue proximal to the stroke site, disrupting circulation to distal areas. Conversely, ischaemic strokes arise from a physical obstruction preventing the movement of blood, leaving areas of the brain starved of oxygen and glucose. The trigger of an ischaemic event is primarily due to the presence of an atherothrombotic clot within the cerebral vasculature, however foreign material and air bubbles can also impede blood flow (Figure 1).
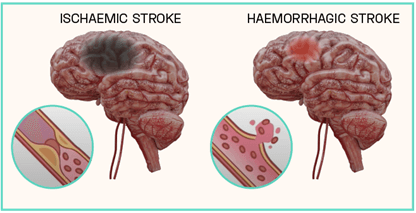
Figure 1: Ischaemic vs Haemorrhagic Strokes
Atherothrombotic clots are generated as a complication of atherosclerosis, a vascular disease characterised by the build-up of hardened plaque (atheroma) within the arterial wall. Various stimuli within the vessel lumen, including high blood pressure and inflammation, enhance the instability of atheromas, predisposing them to rupture. When a rupture occurs, the content of the plaque spills across the vessel lumen and triggers an inappropriate haemostasis response known as thrombosis. Thrombosis favours the coagulation of the blood, resulting in the formation of a blood clot (atherothrombotic clot) which occludes the artery, significantly limiting blood flow.
In this review, we will look closely at the primary cause of ischaemic strokes and atherosclerosis within the cerebral vascular system.
The Cerebral Vasculature and the Brain
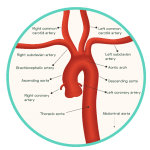
Figure 2: The Aortic Arch and Cerebral Blood Supply
Oxygenated blood is transported to the brain by two pairs of artery branches that extend from the arch of the heart’s aorta: the common carotid arteries and the subclavian arteries. Both sets of arteries ascend upwards within either side of the neck, each bifurcating along their path towards the brain. The subclavian arteries give rise to vertebral arteries at the level of the lower neck, arteries which converge together to form the posterior cerebral circulation. In contrast, the common carotid arteries give rise to the external and internal carotid arteries around the level of the thyroid gland. The internal carotid arteries form the anterior circulation of the brain, while the external carotid arteries branch numerously to perfuse the more superficial structures of the head, such as the scalp.
The intracerebral arterial system supplies the brain with oxygenated blood, transported directly from the heart. This system can be partitioned into an anterior and posterior circulation, supplied by the internal carotid and vestibular arteries, respectively, and their branches (Figure 2). These vessels are structurally adapted to ensure a continuous and sufficient perfusion of oxygen to the brain is maintained at all times. Without a supply of >10 mL/100 g of blood per minute, the neuronal cells of the brain cannot survive and undergo cellular death within minutes. Once this has occurred, permanent brain damage is unavoidable.
With each rhythmic pump, oxygenated blood is forced out from the heart’s left ventricle under high pressure, propelling enough blood forward within the arterial system to reach all metabolically active tissue targets. In the aorta, the body’s largest artery and direct branch of the heart, blood travels at a high velocity of 30 cm/sec. While pressure subsequently reduces downstream, the conducting and distributing cerebral arteries (Figure 3) are continuously subjected to the mechanical effects of high-pressure flow, making them susceptible to vascular inflammation, the critical causative factor in atherosclerosis.
Derived from the Ancient Greek ‘athere’ and ‘sclerosis’ atherosclerosis directly translates to ‘porridge hardening’, relating to its gloopy yellowed visual appearance within the vessel. It progresses from the build-up of solidified lipid and fat deposits within the inner lining of larger arteries, to vessels becoming stiff and constricting in size, compromising their normal function. This process can impact conducting and distributing arteries through the body, however its development within the intracerebral arteries is particular detrimental as it is the biggest risk factor for a
cerebral infarction (cerebral tissue death), more commonly known as an ischaemic stroke. During the early and moderate stages of intracerebral atherosclerosis arterial stiffening itself can inflict mild signs of cerebral hypoperfusion, however the most harmful effects on the brain are seen during the advanced stages of the disease. At these later stages the development of an atherothrombotic clot may occur, which can block almost all of blood flow to the brain and cause tissue necrosis.
Unlike other organs, including the liver and the skin, the brain is incapable of tissue regeneration. A severe loss of oxygenated blood perfusion leads to volumetric tissue loss that is eventually visualised as cavitation. Irretrievable injury to the brain’s neuronal network causes functional areas of this organ to lose the ability to operate, leading to disability. This severely impacts patients’ quality of life and often requires long-term support, both physically and financially. Extensive tissue necrosis or necrosis occurring in regions of the brain controlling basic survival or non-voluntary actions, such as breathing, can lead to sudden death. Infarctions not only lead to immediate damage, but survivors are prone to suffer other degenerative conditions including Parkinson’s disease. Maintaining vascular homeostasis and preventing inflammation in the arterial vessels of the brain is thus fundamental to supporting the normal metabolic activity of brain cells and preventing life threatening damage. Despite modern medicine providing doctors with various diagnostic techniques which help successfully identify the presence of this vascular deterioration, understanding the pathophysiology of atherosclerosis has become a focal point of research since this disease is still attributed to cause one in two deaths in western society.
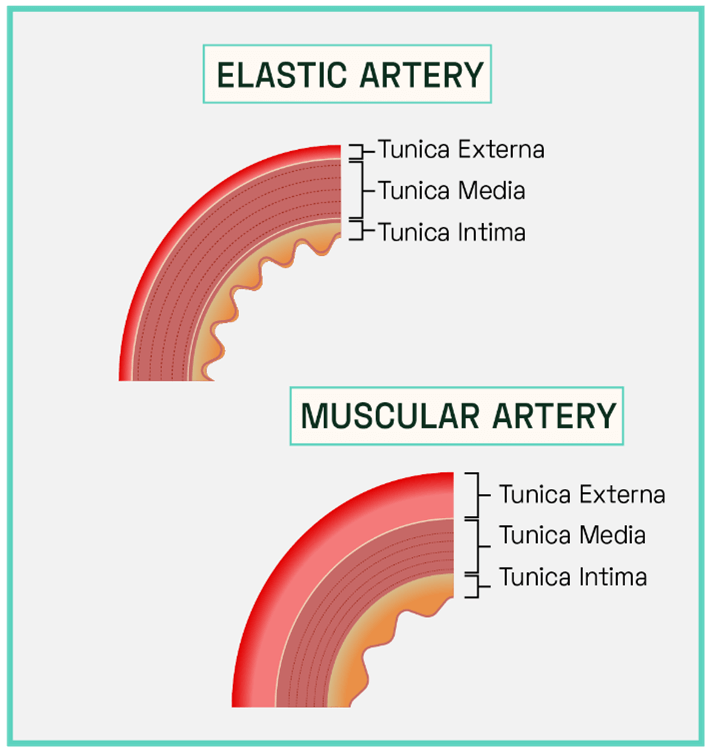
Figure 3: Structure of Conducting and Distributing Cerebral Arteries
The conducting arteries (or elastic arteries) have a thick tunica media, high in elastin. These vessels are direct branches of the heart’s aorta and include the carotid and subclavian arteries. A high ratio of elastin filaments enables these vessels to efficiently transport blood directly away from the heart, allowing for rapid luminal expansion during systole (heart contraction) which lowers vascular resistance and allows the transport of large volumes of blood as the artery lumen expands. Sudden recoil during diastole (heart relaxation) maintains the high pressure gradient as exerted by the heart, propelling the blood forward through the cerebral arterial system.
The distributing arteries, including the internal carotid and vestibular, consist of a thicker tunica media dominated by smooth muscle cells. The luminal diameter of these vessels is much smaller (increased vascular resistance) which prevents damage to the distal arterioles and cerebral tissue. The increased abundance of smooth muscle cells allows for involuntary control of vessel calibre and thus a more precise control of blood flow compared to the conducting arteries.
The Pathophysiology of Atherosclerosis
Classed as a progressive disease, the initiation of the atherosclerotic process, atherogenesis, is seen decades prior to the presentation of any detectable clinical symptoms. Early signs of atherosclerosis can be seen in children, with evidence suggesting they may even appear in new-born babies. For most patients, this disease poses no great threat to survival until much later in life.
This long-term condition is also incredibly complex in nature, with its pathophysiology still being mapped. However, it is broadly divided into five main steps: 1) Endothelium activation; 2) LDL infiltration and oxidation; 3) Foam cell formation and the fatty streak; 4) Vascular remodelling; and 5) Plaque formation. We will now explore each phase of atherogenesis and its pathological link to ischaemic strokes.
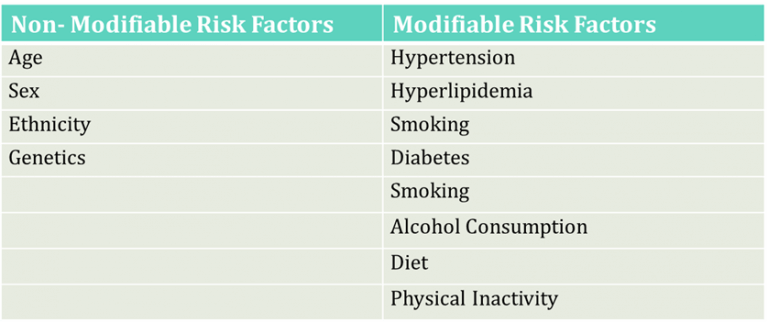
Table 1: Risk Factors for Atherosclerosis
1) Endothelium Dysfunction
In SCIENTIFIC COMMUNIQUE V it was explained that a single layer of endothelial cells constitutes the inner lining (tunica intima) of all arteries, including the intracerebral arteries. These cells are constantly synthesising various vascular agents which help alter vessel tone and regulate vascular immune responses, actions which help preserve vascular health. It is now understood that instigation of atherogenesis follows damage to this inner lining, leading to a loss of normal endothelium functioning, known as endothelium dysfunction (or activation). Endothelium dysfunction is defined as the
characterised shift of these cells towards a proinflammatory, prothrombic and proconstrictive state. One of the most notable changes that occurs within these cells is the inability to synthesise and release nitric oxide (NO), a key signalling molecule involved in vascular maintenance (see SCIENTIFIC COMMUNIQUE V).
Endothelium dysfunction will more often result from the synergistic effect of multiple factors acting upon these cells to induce disruption. Once it occurs, both these cells and underlying vascular tissue become vulnerable to further damage. A complete consensus towards what factors first initiate endothelium dysfunction is yet to be reached in the medical community, however this review will discuss the prevailing hypothesis.
Studies have shown the development of atherosclerosis occurring in predictable regions along the arterial system, forming in focal areas frequently subjected to turbulent blood flow. This includes sites of high curvature and vessel bifurcation. In the intracerebral arterial system, for example, atherosclerotic lesions are most commonly found within the carotid artery at the point where it branches into the internal carotid artery. The ‘haemodynamic hypothesis’ suggests the frictional force within these regions is substantially higher, which disturbs the endothelial glycocalyx layer (see SCIENTIFIC COMMUNIQUE V). Intact, this layer operates as an additional supportive structure to the endothelium, helping modulate vascular permeability and cell adhesion amongst other functions (Figure 4). A large role of the glycocalyx layer is to also modulate the oxidative stress of the endothelial cells. Components of the glycocalyx layer bind to the antioxidant superoxide dismutase (SOD), which in turn ‘mops up’ excess superoxide anions, a type of reactive oxygen species (ROS). Shedding of the glycocalyx layer results in rising levels of ROS within the endothelial cells, which in turn limits NO bioavailability (Figure 4). NO is a potent inhibitor of the transcription factor nuclear factor kappa B (NF‐κB) as it stabilises its inhibitor molecule IκBα. In the absence of IκBα, NF-κB is activated and serves as the master coordinator of the inflammatory response, as it is able to provoke the transcription of an array of pro-inflammatory cytokines (signalling molecules). This results in the enhanced expression of cell adhesion molecules on the surface of the endothelium and the secretion of diverse inflammatory mediators. Endothelial dysfunction has begun. This is just one effect of glycocalyx perturbation. Due to its vast vasculo-protective role, disruption to the glycocalyx layer has many detrimental effects on the adjacent endothelium, all of which cause oxidative stress, inhibition of NO, cytokine production, and enhanced endothelium permeability.
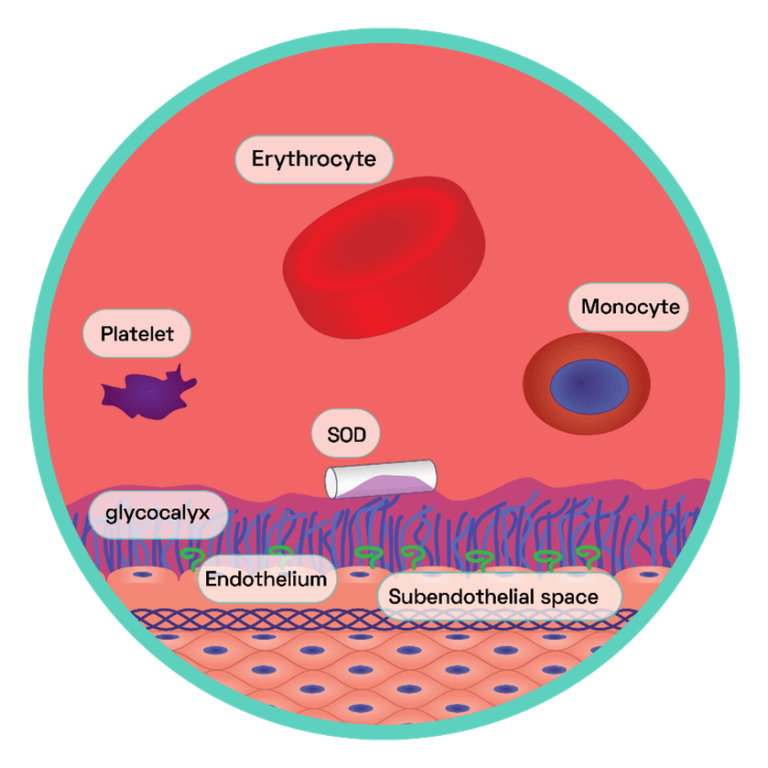
Figure 4: The Endothelial Glycocalyx Layer
This layer is the first primary barrier of carbohydrate rich macromolecules which protrudes into the lumen of the vessel, connected to the apical surface of the endothelium via proteoglycans and glycoproteins. The endothelial glycocalyx regulates NO bioavailability by harbouring extracellular SOD, which maintains low endothelial oxidative stress. This generates oxidative stress within the cells. NO reacts with the surplus of superoxide anions and in doing so forms the strong oxidant peroxynitrite (ONOO−), which is capable of interacting with, and consequently damaging, various cellular components including lipids, proteins and even DNA. One particular protein susceptible to oxidative damage is endothelium nitric oxide synthase (eNOS) the enzyme responsible for NO synthesis. NO promotes vasodilation, anti-adhesion and depresses the expression of pro-inflammatory cytokines (see SCIENTIFIC COMMUNIQUE V).
The endothelium expresses basal levels of cell surface adhesion molecules on its surface, which are upregulated following pro-inflammatory cytokine stimulation. Under normal conditions, the dimension of the glycocalyx by far exceeds that of adhesion molecules thus inhibiting the binding of leukocytes and platelets.
The net charge of this layer serves as a physical and electromagnetic barrier for macromolecules, including plasma proteins and lipoproteins, prohibiting their transmigration across the vascular wall.
2) Low Density Lipoproteins (LDLs) and Oxidation
The endothelium glycocalyx layer also acts as a lubricant by repelling constituents of the blood, preventing their abnormal adhesion to the blood vessels. The loss of this layer means vascular resistance slowly rises as specific molecules that were once deterred from vessel wall interaction can now bind to the endothelium.
Cholesterol is one example of this, a lipophilic molecule whose presence is found in all cells. It has multiple functions within our body, including involvement in hormone synthesis and controlling cell fluidity. Due to their hydrophobic (water repelling) nature, cholesterol molecules are transported to and from various tissues via the blood inside lipoproteins. These are complex particles consisting of a hydrophobic core (cholesterol esters and triglycerides) surrounded by a monolayered hydrophilic (having a strong affinity to water) coat, phospholipids and apolipoproteins. Certain classes of lipoproteins are known contributors to atherosclerosis. Pro-atherogenic lipoproteins include the low density lipoproteins (LDLs) which carry roughly 70% of all serum cholesterol from the liver to target peripheral tissues such as the adrenal glands. The apolipoprotein ligand, apoliprotein B- 100 (apo B-100), allows LDLs to interact with surface receptors on target tissues which subsequently internalise this particle where cholesterol is then extracted and utilised. However, small dense LDLs have a lower affinity for these specific receptors. The apo B-100 ligand is also prone to modifications that further depresses the particle’s clearance from the circulatory system. These types of lipoproteins therefore remain trapped in the blood for prolonged periods of time, increasing their chance of endothelium interaction. As a general rule, these types of lipoproteins are known as ‘bad’ cholesterol.
Injury to the glycocalyx facilitates increases LDL transcytosis across the endothelium where these particles are then deposited in the subendothelial space of the intima. The presence of proteoglycans in the subendothelial extracellular matrix contributes to the retention of these lipids. As LDLs accumulate in the arterial wall, they are rapidly oxidised by ROS molecules, such as superoxide anions released by the overlying dysregulated endothelium. While native LDLs are not associated with damage, in their oxidised form (oxLDL) these particles further induce endothelium dysfunction. This is because structural modifications induced by oxidation allow oxLDL to bind to endothelial surface receptors known as lectin-like receptors (LOX-1). This stimulates the activity of an enzyme complex called nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which generate high levels of ROS, amplifying endothelium oxidative stress and inducing a more severe level of cellular dysfunction as NO bioavailability is further reduced. The surplus of ROS also oxidises more LDLs as they migrate across the vascular wall. With the breakdown of the glycocalyx layer far exceeding its resynthesis, given time, the concentration of subendothelial LDLs becomes uncontrollably high, leaving an aggregated mass of oxidised cholesterol filled particles which lies beneath a now very dysfunctional endothelium. A cycle of transmigration, accumulation and oxidation occurs repeatedly.
3) Foam Cell Formation and the Fatty Streak
The uncontrollable build-up of oxLDLs is the causative force which leads to the next stage of atherogenesis, the development of the fatty streak. The fatty streak is the first grossly visible lesion to form, generated by an accumulation of coalescing oxLDLs and immune cells. They appear yellow-white in colour and are commonly observed in early childhood. At this stage, the body’s own protective inflammatory response has been initiated in response to heightened endothelium dysfunction to help circumvent the damage caused by oxLDL.
A mediator of the pro-inflammatory response is monocyte chemoattractant protein-1 (CCL2). This is a potent chemokine (a type of cytokine) upregulated in dysfunctional endothelial cells following increased LOX-1 signalling and NF‐κB activation. CCL2 is central to the recruitment and activation of a type of blood circulating leukocyte (white blood cell), the monocytes. The release of CCL2 from the abnormal endothelium cells attracts and guides these immune cells into the subendothelial space of the vessel, using the presence of upregulated endothelial adhesion molecules to facilitate their transmigration. The highly oxidative microenvironment of the subendothelial space results in monocyte differentiation into macrophages which can now begin to remove oxLDL via phagocytosis. The body is therefore recognising the build-up of oxLDL as a foreign harmful substance, similar to that of a pathogen, and removing it as such. Though this initially appears beneficial, as the concentration of dangerous oxLDLs in the vessel decreases following uptake, the continuous ingestion of modified LDLs can instead overload the macrophages and also excel the inflammatory response. The interaction between oxLDL and the macrophage receptors induces the secretion of more chemokines, encouraging further infiltration of circulating monocytes and other inflammatory cells, on top of the signals produced by the activated endothelium. Macrophages additionally secrete oxidants, which oxidises further LDLs. What results is the continuous movement of leukocyte derived cells into the heart of the vascular dysfunction, with the pool of oxLDL ever growing in size. Based on current evidence, the amassing of free cholesterol in macrophages is now understood to impair their normal function as excessive free cholesterol hampers the activity of intracellular cholesterol hydrolysing enzymes. This results in the build-up of cytosolic cholesterol droplets, giving the cells a foamy appearance microscopically, which denotes these lipid-laden macrophages foam cells (Figure 5). Eventually, the enlargement of the foam cell population alongside the infiltration of other immune cells results in the development of the fatty streak.
The fatty streak development is the longest asymptomatic phase of atherosclerosis, with disease regression highly possible, assuming early interventions such as improvements in lifestyle are adopted by the patient. Reverse cholesterol transport is a biological pathway that enables the efflux of accumulated cholesterol from macrophages by transporters and/or passive diffusion where they are then collected extracellularly by alternative lipoproteins such as the high-density lipoproteins (HDLs). HDLs deposit excess cholesterol molecules in the liver where they are used appropriately and therefore removed from the vessel wall. The main aim of lifestyle modifications involve both lowering serum LDL concentrations whilst increasing the number of HDLs in the blood. Changes in a patient’s diet and increased physical activity are known factors that can help rebalance these ratios, with the resulting outcomes limiting fatty streak development. Failing regression, the fatty streak can progress into a threatening fibroinflammatory lipid plaque. It is this plaque that has the potential to inaugurate an ischaemic stroke, as plaques encourage luminal narrowing and contribute to artery blockage. Fatty streaks are hence referred to as the precursor lesions of ischaemia.
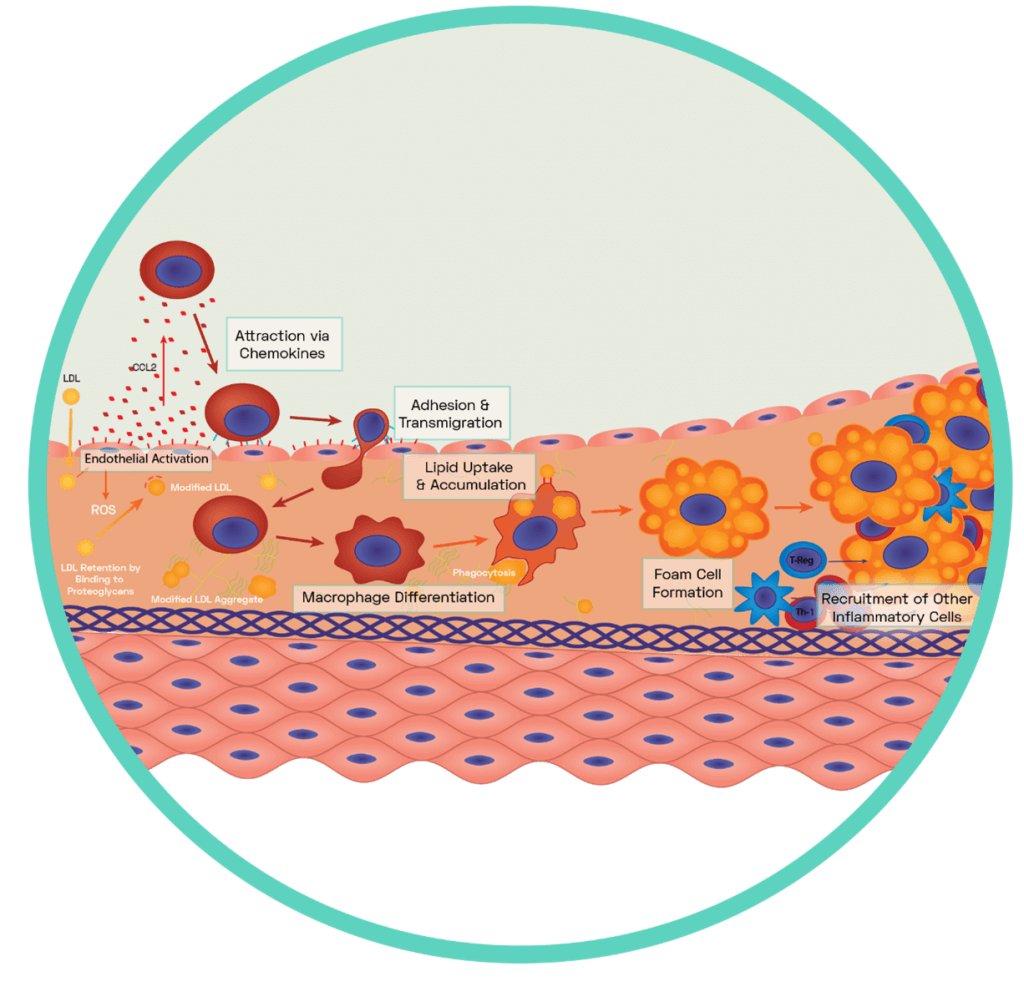
Figure 5: Formation of the Fatty Streak
A dysfunctional endothelium releases a variety of pro-inflammatory chemokines including the potent CCL2 which interacts with blood borne monocytes to guide them to the surface of the dysfunctional endothelium. Via the expression of complimentary ligands, monocytes bind to the exposed endothelial adhesion molecules and transmigration occurs across endothelial cell junctions (see SCIENTIFIC COMMUNIQUE V). Some of these monocytes differentiate into macrophages while others differentiate into dendritic cells. Macrophages display scavenger receptors (i.e SR-A, LOX-1 and CD36) to which apoB-100 binds. This interaction aids the uptake and therefore removal of oxLDL via phagocytosis. It also induces the secretion of more chemokines which encourages further infiltration of circulating monocytes with macrophage secretion of myeloperoxidase, oxidising more LDLs. Once internalised by the macrophages, oxLDL molecules are broken down into smaller and more manageable components in specific compartments called lysosomes. This has an enhancing effect as components of oxLDL are used to increase the expression of more scavenger receptors for further uptake. The enzyme lysosomal acid lipase (LAL) is found within macrophage lysosomes and hydrolyses cholesteryl esters derived from oxLDL into free cholesterol. However, the build-up of free cholesterol impairs the macrophages as excessive free cholesterol hampers the activity of LAL. Cytosolic cholesteryl ester droplets build, causing foam cell formation.
4) Vascular Remodelling and Plaque Formation
Vascular remodelling – changes to the structure of vessels – is the sequential pathological event following lesion formation in the atherogenesis pathway. The development of fatty streak lesions into this disease phase is associated with a heightened clinical significance, as reorganisation of the intracerebral artery wall is induced. The control of blood flow thus digresses, which can be seen as a slight raise in blood pressure.
Under normal conditions, the vascular smooth muscle cells (VSMCs) are in a quiescent state, located predominantly within the tunica media layer of the vessel. The further release of cytokines such as transforming growth factor-β (TGF-β), fibroblast growth factors (FGF) and matrix metalloproteinases (MMPs) from the foam cells causes a radical change to the behaviour of VSMCs.
VSMCs have two major functions. The first was explained in detail in SCIENTIFIC COMMUNIQUE V and involves controlling vascular tone. Their second function involves synthesising and degrading the surrounding extracellular matrix, a mesh heterogenous network of non-cellular connective tissue. Cytokine signalling from the lesion causes the local migration of VSMCs into the intima. This is followed by cell proliferation (multiplication). VSMCs aggregate on the luminal side of the lesion and secrete numerous extracellular matrix components which include collagen and proteoglycans. These molecules form a fibrous cap over the mound of foam cells which is believed to be an attempt to sequester the cumulus of foam cells, preventing communication between lumen of the vessel and this area of disease. However, in doing so, the fibrous cap stabilises the fatty streak. Once fibrous reinforcement of the plaque occurs, the likelihood of regression of the fatty streak is significantly reduced. The regulatory T cells make up one of the many immune cells that have successfully migrated from the blood into the vessel wall alongside the macrophages in response to endothelium dysfunction. Both the regulatory T cell and macrophages release TGF-β, which stimulates the smooth muscle cells to secrete collagen, further reinforcing cap durability. As this process continues the cap thickens, measuring around 1 to 2mm, which causes a slight outward protrusion of the intima as the developing lesion is compressed against the media.
At the same time, a subset of VSMCs collects within the fatty streak alongside macrophages and other inflammatory cells. Some of these cells take on a macrophage-like role, engulfing more oxLDL as the process of LDL transcytosis continues. The competency of this process is questionable as the generation of lipid lakes is seen, suggesting VSMCs are unsuccessful in neutralising oxLDL accumulation. Engorging oxLDL may instead promote cell death, where these VSMCs discharge any ingested lipids and their internal components into the foam cell mass. Other smooth muscle cells will instead display osteogenic characteristics, releasing calcium into the surrounding area. Collectively, this changes the composition of the fatty streak, and alongside the addition of the fibrous cap, form the atheroma plaque.
With time, some foam cells undergo a less common form of cell death known as necroptosis (programmed necrosis) as well as apoptosis. This is caused by stimuli within the plaque including high concentrations of oxidative stress, released by the surrounding endothelium and immune cells. A small necrotic core is left at the heart of the plaque, contaminated with proinflammatory macrophage debris, though the presence of regulatory T cells limits its further development (Figure 5). The regulatory T cells are unique as they innately supress the actions of other immune cells through synthesising and secreting anti-inflammatory cytokines such as interleukin- 10 (IL-10) and, of course, TGF-β. At the adequate concentrations, IL-10 and TGF-β are thought to contribute to the retardation of atheroma advancement by contradicting other biological actions of the pro-inflammatory signals.
While the majority of disease impact is seen within the subendothelial space, structural changes can now also be observed in the tunica media as the disease progresses. Macrophage derived MMPs contribute to the fragmentation of the elastin fibres that reside in the middle layer of the vessel wall. The presence of elastin allows for reversible extensibility during cyclic loading of the cardiac cycle. This enables the conductance of sufficient blood volumes at appropriate pressures towards metabolic tissue, including the brain. Calcium released by the osteogenic VSMCs also diffuses across into the media which consequently causes wall calcification. This stiffens the arteries and, alongside limited NO bioavailability, atherogenic vessels become more permanently contracted. These processes precede raised blood pressure, which not only has the potential to damage the systems smaller and more distal blood vessels (arterioles and capillaries) but increase the workload of the heart. Since narrow and stiff arteries are less efficient in transporting blood, the heart is forced to work harder to sustain normal tissue perfusion, resulting in myocardial atrophy.
5) Plaque formation: Stable vs Vulnerable Plaques
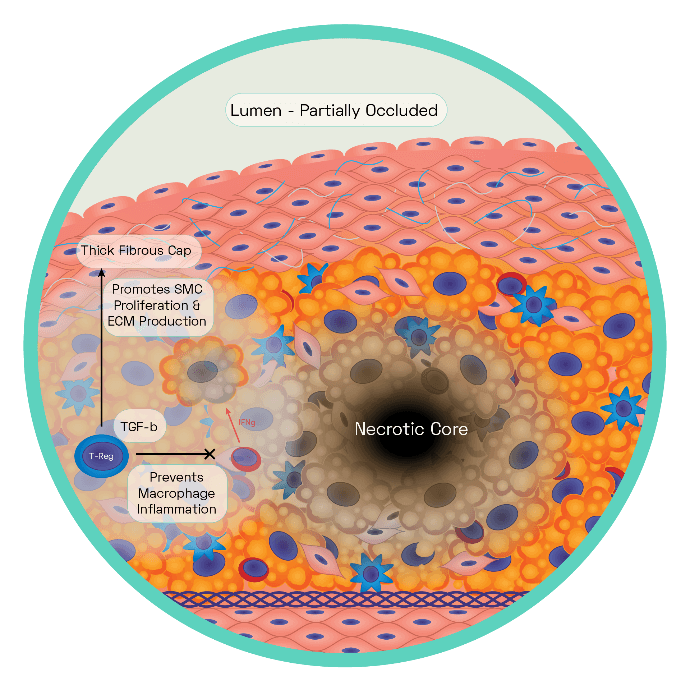
Figure 6: The Stable Plaque
The stable plaque houses a thick fibrous cap composed of layers of VSMC ensconced in a substantial extracellular matrix network of collagen, proteoglycans, and elastin. This is acting as an effective barrier between the lesions prothrombotic factors and the blood, inhibiting thrombosis and therefore ischaemic stroke.
Regulatory T cells and macrophages secrete TGF-β which stimulates VSMC collagen synthesis, with the cap growing in size as a result. Regulatory T cells inhibit T helper 1 cells production of interferon gamma (IFNg). Increased TGF-β and decreased IFNg reduce the proinflammatory macrophage phenotype leading to reduced cell death, effective efferocytosis. Stable plaques are characterised by small necrotic cores and thick plaque caps.
By this stage, the cell volume within the intima has increased considerably, owing to the continuous build-up of foam cells, proliferating VSMCs smooth muscle cells and necrotic tissue. Limited room for growth within the subendothelial space forces the plaque to further encroach on the territory of the tunica media as it expands outward (Figure 6). Eventually the plaque is forced to mature inwards, pressing into the lumen of the vessel. Partial occlusion of the vessel’s diameter can be seen, with the presence of clinical symptoms at this stage variable, and dependant on the extent of ingrowth. In extreme cases cerebral stenosis may be observed, with severe narrowing leading to a lack of sufficient blood flow to areas of the brain, resulting in vertigo, sudden headaches and numbness.
While the plaque is large, it remains stable. Stable atheromas may remain dormant in the arterial wall for long periods of time. However, despite their name, they can eventually rupture. A shift in the equilibrium between the activity of anti-inflammatory and pro-inflammatory immune cells can trigger the development of a larger necrotic core. Stable plaques house only a minimal number of necrotic cells, yet the decreased expression of IL-10 and TGF-β, coupled with defective clearance of cellular waste (efferocytosis), sees the number of necrotic cells dramatically increase. It has been hypothesised that nutrient deprivation may potentially accelerate lipid laden macrophage death. This in turn releases more pro-inflammatory mediators as part of the necrotic process which cycles to further induce cell death of neighbouring foam cells. Faulty removal of dead cells and their harmful debris (efferocytosis) appears to result in a secondary round of necrosis, consisting of more leakage of oxidative and inflammatory components. This cycle of unresolving inflammation and cellular death eventually starts to have an effect on the smooth muscle cells and fibrous cap that encompasses the topical surface of the plaque. Apoptosis of the smooth muscle cells follows the intensification of the necrotic core.
Since these cells are responsible for the secretion of extracellular elements that make up the atheroma’s cap, the production of fibrous components slows. With TGF-β expression dampened, synthesis of collagen decelerates from the remaining smooth muscle cells. Pro-inflammatory agents such as MMPs work to degrade the fibrous cap further. Eventually the cap becomes extremely thin (< 1 mm thick), leaving behind a vulnerable plaque.
In the absence of a thick, stiff, protective top layer, the unstable plaque is prone to fracturing under the continuous shear and cyclic stresses. Decreased compliance in a diseased artery leads to disturbed turbulent flow patterns from the blood. Along with expression of MMPs, ROS and other damaging cytokines from the necrotic plaque, endothelium erosion occurs. Cell to cell junctions are degraded, resulting in high vascular permeability which is only enhanced by the increased rates of endothelial apoptosis. The site most vulnerable to plaque attrition is known as the ‘shoulder’, a region of the diseased vessel characterised by few smooth muscle cells and a high concentration of macrophages. Eventually the plaque is directly exposed to the force of the blood which leads to plaque fissures that trigger a rupture. The contents of the plaque spills out of the vessel which is rapidly followed by the abnormal activation and accumulation of haemostatic factors. Oxygenated blood is rich in platelets which, alongside erythrocytes (red blood cells), attempt to plug the site of injury to inhibit further damage and repair the vessel. However, the effects of aggregation and coagulation result in the formation of an obscuring mass of cells that encompasses the rupture (an atherothrombotic clot or thrombus). The artery constricts, adding to luminal occlusion alongside the clot (Figure 7).
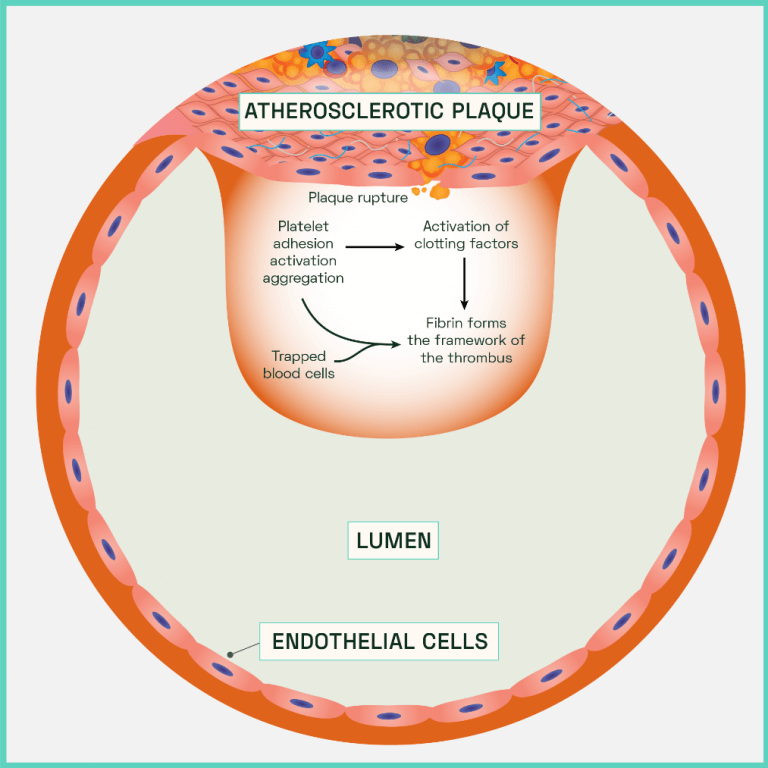
Figure 7: Atherothrombotic clot (thrombus) formation
However, plaque rupture is often a recurring matter and is not always initially lethal. In cases where smooth muscle cells are still functional, these cells quickly accumulate at the rupture where they then secrete fibrous components across the damage vessel and necrotic debris to temporarily impede the thrombosis response. However, this barrier is weak and eventually breaching occurs. Successful plaque repair relies on a sufficient number of operational smooth muscle cells to be retained within and surrounding the plaque. Ultimately, signals from the necrotic core induce death and cellular senescence of the smooth muscle cells which depletes their ability to induce repair.
Atherosclerosis and Ischaemic Stroke
A plaque rupture in an intracerebral artery which is superimposed by thrombosis can result in an ischaemic stroke if enough blood is prevented from reaching the brain. At times, the obstruction formed is not large enough to significantly occlude the entirety of the diseased artery’s lumen and an adequate volume of oxygenated blood may still pass to meet the metabolic needs of the brain. However, the movement of passing blood across the surface of the atherothrombotic clot increases the risk of thromboembolus transformation. This occurs when pieces of the abnormal clot become dislodged from the original mass. The now mobile debris (embolus) migrates within the blood stream, reaching distal cerebral arteries. With these vessels dramatically narrower in size, an embolus risks become trapped further within the cerebral arterial system. Similar to the primary atherothrombotic clot, an obstructing embolus triggers ischaemic conditions within the brain tissue as blood perfusion is prevented.
Summary
Considering the pathogenesis of atherosclerosis, a combination of lifestyle modifications and pharmacological interventions have been the mainstays of treatment to reduce disease progression, although in contemporary medicine particular attention has been paid to treatments that target the final and most detrimental stage of atherogenesis in a means to directly prevent the occurrence of an ischaemic stroke.
Despite this, atherosclerosis still remains the leading cause of mortality and disability worldwide, with figures rapidly rising in developing counties. In addition, aging populations and elevations in care costs have led to some countries predicting the economic burden of diseases secondary to atherogenesis, such as strokes, will increase by nearly 200% between 2015 and 2035. This therefore demonstrates that despite current management of risk factors and progresses in treatments for atherosclerosis, improvements are still needed to curtail the occurrence of ischaemic stroke which comprise most stroke cases. In a following SCIENTIFIC COMMUNIQUÉ we will discuss the current therapies for both atherosclerosis and ischaemic strokes, reviewing the effectiveness of existing treatments.
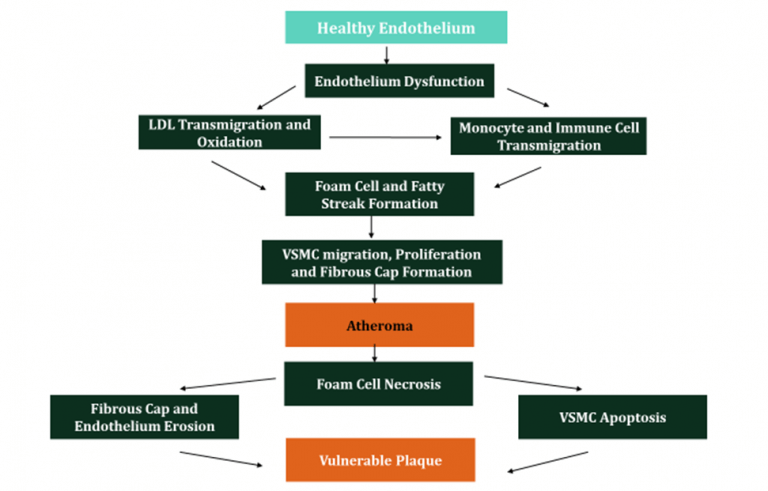
Figure 8: Summary of Atherosclerosis
Glossary
| Term | Definition |
| Aorta | The main and largest artery in the human body, originating from the left ventricle of the heart. |
| Apolipoproteins | An amphipathic molecule capable of interacting with the lipids of the lipoprotein core and the aqueous environment of the plasma. |
| Apoliprotein B- 100 (apo B-100) | A class of apolipoproteins. Apo B-100 contains a binding domain for receptors including LDL receptors and are abundantly associated with LDLs. |
| Apoptosis | Programmed cell death. |
| Atherosclerosis | A dangerous condition where arteries become clogged with fatty substances called plaques, or atheroma. |
| Atherogenesis | A process that results in the disease atherosclerosis. It is the progressive thickening and hardening of the walls of arteries as a result of fat deposits on their inner lining. |
| Atheroma | A fatty deposit in the inner lining (intima) of an artery, resulting from atherosclerosis. Also called an atherosclerotic plaque. |
| Atherothrombosis | Atherosclerotic plaque disruption with superimposed thrombosis. |
| Basal lamina | Also known as the basement membrane, is a specialised form of extracellular matrix separating the vascular endothelium and subendothelial space. |
| Chemokines | A family of chemoattractant cytokines secreted by cells to induce directed chemotaxis in nearby responsive cells. An example includes the signalling molecule CCL2. |
| Cholesterol | Cholesterol is a waxy, fat-like substance that occurs naturally in the body and is found in all cells. |
| Conducting arteries | Otherwise referenced to as the elastic arteries, they are large arteries with many collagen and elastin filaments in the tunica media, which gives them the ability to stretch in response to each pulse. Includes the aorta and its direct branches. |
| Cytokines | A large group of proteins, peptides or glycoproteins that are secreted by specific cells of immune system. They mediate the body’s immunity and inflammation response. |
| Distributing arteries | Otherwise referenced as muscular arteries, these are medium sized arteries that transport blood from an elastic artery and branch into resistance vessels. Many layers of smooth muscle cells comprise its tunica media. Examples include the internal carotid arteries. |
| Efferocytosis | The process of dead cell removal, which involves phagocyte recruitment, dying cell recognition, and engulfment. |
| Endothelial cells | Flattened cell type that forms a sheet (the endothelium). |
| Endothelium | A layer of flattened cells (endothelial cells) that line the blood vessels and lymph vessels of the body. |
| Endothelium activation | See endothelium dysfunction. |
| Endothelium dysfunction | A proinflammatory and procoagulant state of the endothelial cells lining the lumen of blood vessels. It is mostly characterised by an increase in interactions with white blood cells (leukocytes), and is associated with the early states of atherosclerosis. |
| Endothelium nitric oxide synthase (eNOS) | Endothelium nitric oxide synthase, constitutively expressed in the endothelium synthesising NO. |
| Fatty streak | The first grossly visible lesion in the development of atherosclerosis. It is composed of macrophages, LDLs , oxLDLs and other immune cells. |
| Fibrous cap | A distinct layer of connective tissue covering the lipid core of an atherosclerotic plaque. It is composed of smooth muscle cells within a collagenous-proteoglycan matrix. |
| Foam cell | A type of macrophage that localise to fatty deposits on blood vessel walls, where they ingest low-density lipoproteins and become laden with lipids, giving them a foamy appearance. |
| Glycocalyx layer | A carbohydrate-rich layer lining the vascular endothelium. |
| Ischaemia | Blood flow is restricted or reduced in a part of the body. |
| Leukocyte | A general name for all the nucleated blood cells lacking haemoglobin. May also be referred to as white blood cell. |
| Lumen | The inside space of a tubular structure, such as an artery or intestine. |
| Lysosomes | Membrane-bound cell organelle that contains digestive enzymes. |
| Macrophage | Differentiated monocyte that ingests foreign material. |
| Matrix metalloproteinases (MMPs) | Protease enzymes that can break down proteins, such as collagen, that are normally found in the spaces between cells in tissues (i.e., extracellular matrix proteins). |
| Monocyte | A large phagocytic leukocyte. |
| Necroptosis | Programmed form of necrosis, or inflammatory cell death. |
| Necrosis | Uncontrolled cell death that can occur in response to infection, toxins, chemicals, injury, or lack of blood supply. |
| Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase | A membrane-bound enzyme complex that generates high levels of ROS. |
| Nitric Oxide (NO) | A gaseous signalling molecule that is widely used in cell-cell communication. |
| Oxidation | Loss of electrons from an atom. |
| Oxidative stress | An imbalance between reactive oxygen species and antioxidants. |
| Oxidised Low Density Lipoprotein (oxLDL) | Modified form of LDLs. |
| Phagocytosis | The ingestion of bacteria or other material by phagocytes. |
| Platelets | Cell fragment lacking a nucleus that is found abundantly in the blood stream. Helps initiating blood clotting in injured blood vessels. |
| Reactive Oxygen Species (ROS) | Chemically reactive chemical species containing oxygen, such as the superoxide anion or hydrogen peroxide. |
| Regulatory T cells | Cells which have a role in regulating or suppressing other cells in the immune system. |
| Scavenger receptors | A large and diverse superfamily of cell surface receptors. |
| Subendothelial space | The thin layer of connective tissue lying between the endothelium and elastic lamina in the intima of blood vessels. |
| Thrombosis | The formation of a thrombus (blood clot) within a vessel. |
| Tunica externa | Also referred to as the tunica adventitia. The outer layer of blood vessels consisting mainly of supportive collagen fibres. |
| Tunica intima | The innermost layer of a vessel, consisting of a layer of endothelium sitting on top an internal elastic lamina. |
| Tunica media | The middle layer of a blood vessel consisting of smooth muscle cells, elastic tissue, fibroblasts and extracellular matrix. |
| Tunica intima | The innermost layer of a vessel, consisting of a layer of endothelium sitting on top an internal elastic lamina. |
| Vascular homeostasis | The balance between vascular injury and vascular repair. |
| Vascular tone | The level of vessel constriction relative to its maximally dilated state. |
| Vasoconstriction | The narrowing or constriction of the blood vessels, reducing the size of the blood vessel opening (lumen). Blood flow is reduced, and blood pressure increases. |
| Vasodilation | The dilation, or widening, of blood vessels, increasing the size of the vessel opening (lumen). Vasodilation causes increased blood flow through the blood vessels and decreased blood pressure. |
References
Kim, J. S., & Bang, O. Y. (2017). Medical treatment of intracranial atherosclerosis: an update. Journal of stroke, 19(3), 261.
Kojima, Y., Weissman, I. L., & Leeper, N. J. (2017). The role of efferocytosis in atherosclerosis. Circulation, 135(5), 476-489.
Linton, M. F., Yancey, P. G., Davies, S. S., Jerome, W. G., Linton, E. F., Song, W. L., … & Vickers, K. C. (2019). The role of lipids and lipoproteins in atherosclerosis. In Endotext [Internet]. MDText. com, Inc.
Nieuwdorp, M., Meuwese, M. C., Vink, H., Hoekstra, J. B., Kastelein, J. J., & Stroes, E. S. (2005). The endothelial glycocalyx: a potential barrier between health and vascular disease. Current Opinion in Lipidology, 16(5), 507–511.
Patel, A., Berdunov, V., King, D., Quayyum, Z., Wittenberg, R. and Knapp, M., 2017. Current, Future And Avoidable Costs Of Stroke In The UK. [online] Stroke.org.uk.
Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics—2020 update: a report from the American Heart Association external icon. Circulation. 2020;141(9):e139–e596.
Whayne Jr, T. F. (2011). Atherosclerosis: current status of prevention and treatment. The International journal of angiology: official publication of the International College of Angiology, Inc, 20(4), 213.