Xeroderma Pigmentosum
About XP
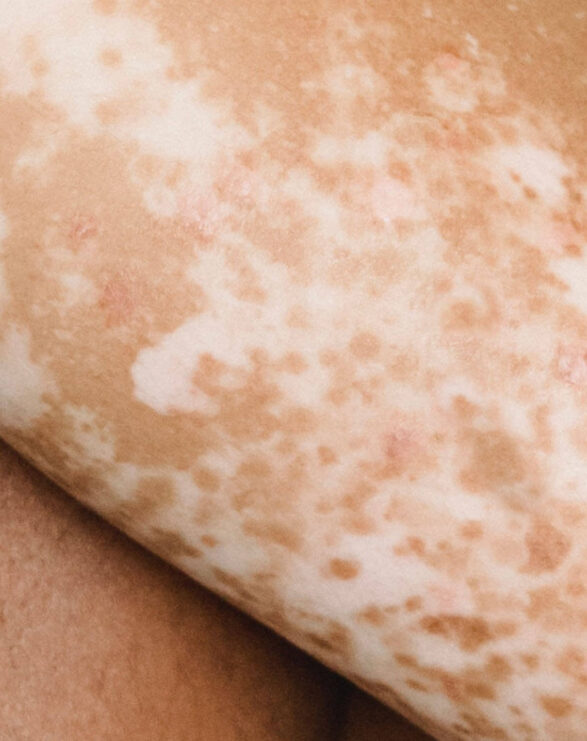
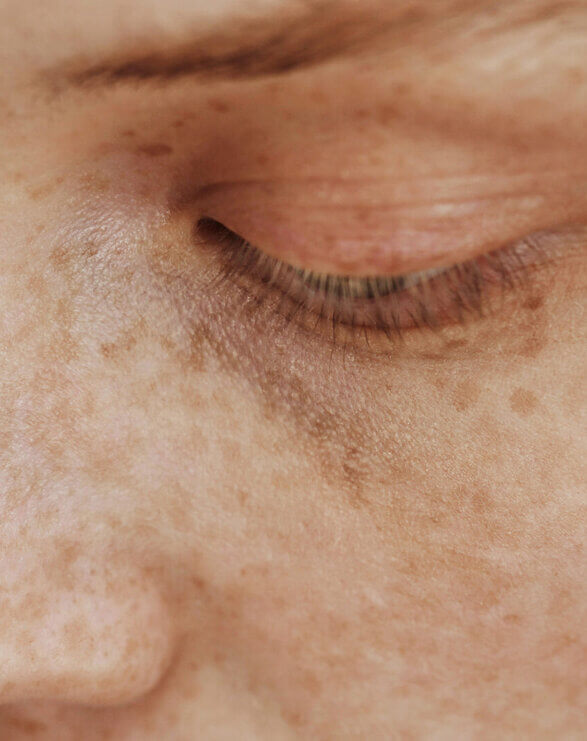
Xeroderma pigmentosum is a debilitating rare disease characterised by hyperkeratinisation, poikiloderma and wider skin tissue damage that is a consequence of irreparable DNA damage. Affected individuals are particularly susceptible to developing actinic keratoses and squamous and basal cell carcinomas due to a genetic defect which hampers the nucleotide excision repair (NER) process. Eye and neurological problems are likely to occur over time, but neurological defects only develop in those with the greatest impairment of DNA repair.
Due to these genetic deficiencies in DNA repair proteins, XP patients are at a 10,000-fold greater risk of developing skin cancers including melanoma, necessitating them to shield from ambient and outdoor light from birth onwards. The extreme rate of these malignancies requires frequent surgical removal that often results in loss of extremities and facial anatomy, such as ears, and eyesight. The median age of survival for XP patients is approximately 30 years.
XP is an autosomal recessive disease, which means an individual with the disease has inherited two copies of the mutated gene (one from each parent) that causes XP. If only one XP gene is inherited, then that person carries the gene but is not affected by it. Having two XP genes causes patients to be incapable of responding to DNA damage, resulting in a range of symptoms.
The symptoms are caused by faulty DNA repair mechanisms in the skin. A defect in nucleotide excision repair (NER) results in an inefficient DNA repair mechanism in XP. Ultraviolet radiation (290 – 400nm) and high energy visible (HEV, 400-600 nm) light from sun exposure damages DNA. As the cells are unable to repair it, the DNA remains damaged, which leads to cell death and the development of cancerous cells.
What is Xeroderma Pigmentosum (XP)?
Learn more about the rare hereditary disease Xeroderma Pigmentosum and how it can be treated.
Clinical & regulatory progress
November 2024
CLINUVEL announced that its XP program will be temporarily suspended. More details are in the announcement here.
January 2023
CLINUVEL released the first results of the Phase II study (CUV156) evaluating afamelanotide in patients with XP. Analyses showed a decrease in ultraviolet (UV) light-induced DNA skin damage following treatment. Read the announcement here.
March 2022
The first xeroderma pigmentosum variant (XP-V) patient to receive afamelanotide treatment in the third active study in the Company’s DNA Repair Program (CUV152) was announced on 30 March 2022.
December 2021
Two xeroderma pigmentosum (XP) patients received afamelanotide treatment in the CUV156 study, part of the Company’s DNA Repair Program. Read the announcement.
October 2021
CLINUVEL announced that it had received the necessary regulatory and ethics committee approvals to commence a new study in XP, CUV156. The first patient, with the XP-C complementation, was treated in the CUV156 study in late October. Read the announcement here.
September 2020
CLINUVEL announced the launch of its novel DNA Repair Development Program to confirm the ability of SCENESSE® (afamelanotide 16mg) to repair damaged DNA and investigate its use as a treatment for xeroderma pigmentosum.
The aim of the development program is to confirm the drug’s ability to regenerate DNA of skin exposed to ultraviolet (UV) damage.
References
Kramer KH, Patronas NJ, Schiffmann R, Brooks BP, Tamura D, DiGiovanna JJ. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Neuroscience. 2007;145(4):1388-1396. doi:10.1016/j.neuroscience.2006.12.020.
Lichon V, Khachemoune A. Xeroderma pigmentosum: beyond skin cancer. J Drugs Dermatol. 2007;6(3):281-288.
Sugasawa, K. Xeroderma pigmentosum genes: functions inside and outside DNA repair. Carcinogenesis. 2008;29(3):455-465. doi:10.1093/carcin/bgm282.
Zahid, S, Brownell, I. Repairing DNA damage in Xeroderma Pigmentosum: T4N5 lotion and gene therapy. J Drugs Dermatol. 2008 Apr;7(4):405-8. PMID: 18459526.
Zeng, L, Quilliet X, Chevallier-Lagente O, Eveno E, Sarasin A, Mezzina M. Retrovirus-mediated gene transfer corrects DNA repair defect of xeroderma pigmentosum cells of complementation groups A, B and C. Gene Ther. 1997;4(10):1077-1084. doi:10.1038/sj.gt.3300495.